Total synthesis of batzelline C and isobatzelline C†
Takashi
Oshiyama
,
Takahito
Satoh
,
Kentaro
Okano
and
Hidetoshi
Tokuyama
*
Graduate School of Pharmaceutical Sciences, Tohoku University, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: tokuyama@mail.pharm.tohoku.ac.jp; Fax: (+81) 22 795-6877; Tel: (+81) 22 795-6887
First published on 20th April 2012
Abstract
The total synthesis of batzelline C and isobatzelline C was accomplished. The synthesis features the construction of the highly substituted pyrrolo[4,3,2-de]quinoline skeleton by a benzyne-mediated cyclization-functionalization sequence.
The pyrroloquinoline alkaloid family, which includes damirones, batzellines, makaluvamines, and discorhabdins, is an important class of natural products since these compounds possess numerous intriguing biological activities.1 Among them, batzellines2 and isobatzellines,3 isolated from the marine sponge Batzella sp., have recently been proven to show cytotoxic activity based on the inhibition of topoisomerase II and, in particular, the latter compound has been found to inhibit HIV-1 envelope-mediated cell fusion with an IC50 of 200 nM level (Fig. 1).4 In addition to these significant biological activities, the highly functionalized pyrrolo[4,3,2-de]quinoline structure has attracted considerable attention from the synthetic organic community and, to date, three total syntheses of batzelline C and isobatzelline C have been reported.5 Herein, we report the total synthesis of batzelline C (1c) and isobatzelline C (2c) featuring a novel construction of functionalized pyrrolo[4,3,2-de]quinolines via benzyne-mediated one-pot cyclization-functionalization sequence.6
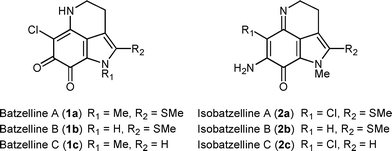 | ||
| Fig. 1 Structures of batzellines A–C and isobatzellines A–C. | ||
Retrosynthetic analysis of batzelline C (1c) and isobatzelline (2c) is shown in Scheme 1. These compounds would be synthesized from the common intermediate 3via oxidation of the aromatic ring. For the construction of the functionalized pyrrolo[4,3,2-de]quinoline skeleton, we designed a one-pot cyclization-chlorination sequence based on the application of the benzyne-mediated one-pot cyclization-arylation sequence (Scheme 2), which was developed in our synthesis of dictyodendrins.7 Thus, treatment of 4-halo-6-methoxyindoline derivative 4 with excess base should provide the corresponding benzyne species 5, which would undergo cyclization by intramolecular attack of the nitrogen nucleophile and chlorination of the resultant arylanion species.
 | ||
| Scheme 1 Retrosynthetic analysis. | ||
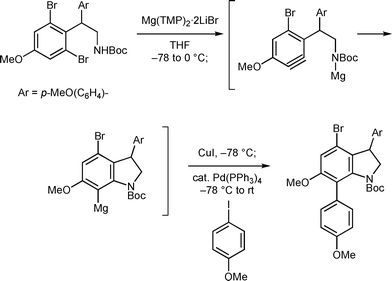
The synthesis commenced with the preparation of the known indoline 6 from the commercially available compound 7 in a 66% overall yield utilizing Buchwald's zirconocene-mediated 4-iodoindoline synthesis using diallylaniline 88 (Scheme 3). The iodomethyl chain at the 3-position of indoline 6 was elongated by conversion to nitrile, LAH reduction, and Boc protection of the resultant primary amine to give Boc carbamate 9. The allyl group was then switched to the ethyloxycarbonyl group by treatment with ethyl chloroformate and sodium iodide to provide carbamate 10.
 | ||
| Scheme 3 Synthesis of precursor for the benzyne-mediated cyclization-chlorination. | ||
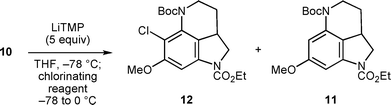
With 4-iodoindoline derivative 10 in hand, we then tested the construction of the pyrrolo[4,3,2-de]quinoline skeleton by the formation of a nitrogen-containing six-membered ring via a benzyne intermediate. We initially explored a suitable base to promote benzyne formation and cyclization. Among a variety of bases tested,9 LiTMP10 gave the best result. Thus, the benzyne generation-cyclization cascade proceeded smoothly to furnish the desired tricyclic compound 11 in 80% yield (Scheme 4).
![Construction of pyrrolo[4,3,2-de]quinoline skeleton.](/image/article/2012/RA/c2ra20604h/c2ra20604h-s4.gif) | ||
| Scheme 4 Construction of pyrrolo[4,3,2-de]quinoline skeleton. | ||
Encouraged by the successful construction of the tricyclic skeleton, we then examined the one-pot cyclization-chlorination sequence by trapping of the aryl anion intermediate with a several chlorinating agents (Table 1). To our disappointment, termination of the reaction with N-chlorosuccinimide (NCS) gave only a minute amount of the desired product 12 associated with the generation of protonated product 11 (Entry 1). Use of TfCl11 was also ineffective (Entry 2). Eventually, we found that the use of C2Cl6 dramatically improved the sequential chlorination reaction and the desired chlorinated compound 12 was obtained in 64% overall yield from 10 (Entry 3).12
Having successfully established the benzyne-mediated cyclization-chlorination sequence for the construction of the chlorosubstituted pyrrolo[4,3,2-de]quinoline skeleton, we then pursued further transformations to synthesize batzelline C (1c) and isobatzelline C (2c) (Scheme 5). First, indoline 12 was converted to indole 13 by DDQ oxidation. Then, Boc and ethyloxycarbonyl groups were sequentially removed in a stepwise manner to give 14. These deprotection steps were crucial for the subsequent smooth oxidation of 14 to iminoquinone derivative 15 using Fremy's salt.5b Thus, reaction of indoles 13 or 16 resulted in slower conversion, possibly due to insufficient electron density of the benzene rings, with the generation of a substantial amount of unidentified side products. Finally, a methyl group was installed at the pyrrole nitrogen and subsequent treatment with boron tribromide affected the demethylation of the methoxy group and spontaneous tautomerization to furnish batzelline C (1c) in 61% over 2 steps. Isobatzelline C (2c) was also synthesized in 53% over 2 steps from compound 17 by treatment with ammonium chloride.5a The physical properties of both synthetic compounds were identical in all aspects to those reported for the natural products.1,2
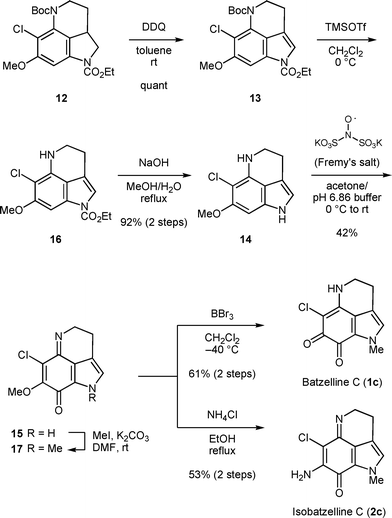 | ||
| Scheme 5 Total synthesis of batzelline C and isobatzelline C. | ||
In conclusion, we have achieved a total synthesis of batzelline C (1c) and isobatzelline C (2c) featuring the newly developed synthesis of a functionalized pyrrolo[4,3,2-de]quinoline skeleton by a benzyne-mediated cyclization-functionalization sequence. This sequential one-pot process enabled us to construct the substituted pyrroloquinoline skeleton in a regiospecific manner, which is superior to the conventional stepwise construction of the tricyclic ring and subsequent functionalization that normally causes regiochemical problems. This strategy would be a powerful tool for the synthesis of not only this class of alkaloids, but also a broad range of functionalized tetrahydroquinoline containing heterocycles.
This work was financially supported by the Ministry of Education, Sports, Science, and Technology, Japan, the KAKENHI, a Grant-in-Aid for Young Scientists (Start-up; 19890014, B; 21790006 and 23790004), the Cabinet Office, Government of Japan through its “Funding Program for Next Generation World-Leading Researchers (LS008), Tohoku University Global COE program ‘International Center of Research and Education for Molecular Complex Chemistry’, Suntory Institute for Bioorganic Research (Sanbor Grant), Nagase Science and Technology Foundation, Astellas Foundation for Research on Metabolic Disorders, Banyu Life Science Foundation International.
References
- (a) D. C. Radisky, E. S. Radisky, L. R. Barrows, B. R. Copp, R. A. Kramer and C. M. Ireland, J. Am. Chem. Soc., 1993, 115, 1632 CrossRef CAS; (b) A. Miyanaga, J. E. Janso, L. McDonald, M. He, H. Liu, L. Barbieri, A. S. Eustáquio, E. N. Fielding, G. T. Carter, P. R. Jensen, X. Feng, M. Leighton, F. E. Koehn and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 13311 CrossRef CAS.
- S. Sakemi, H. H. Sun, C. W. Jefford and G. Bernardinelli, Tetrahedron Lett., 1989, 30, 2517 CrossRef CAS.
- H. H. Sun, S. Sakemi, N. Burres and P. McCarthy, J. Org. Chem., 1990, 55, 4964 CrossRef CAS.
- L. C. Chang, S. Otero-Quintero, J. A. Hooper and C. A. Bewley, J. Nat. Prod., 2002, 65, 776 CrossRef CAS.
- (a) X. L. Tao, S. Nishiyama and S. Yamamura, Chem. Lett., 1991, 1785 CrossRef CAS; (b) F. Yamada, S. Hamabuchi, A. Shimizu and M. Somei, Heterocycles, 1995, 41, 1905 CrossRef CAS; (c) D. Roberts and J. A. Joule, J. Org. Chem., 1997, 62, 568 CrossRef CAS.
- (a) Iwao and co-workers reported a benzyne-mediated approach for makaluvamines using indole substrate and lithium isopropylcyclohexylamide. In this report, no cyclization-functionalization sequence was described: M. Iwao, O. Motoi, T. Fukuda and F. Ishibashi, Tetrahedron, 1998, 54, 8999 CrossRef CAS . In the reported double functionalization of benzyne with a nitrogen nucleophile, a strong base such as s-BuLi6b or intramolecular trapping6c–6f of the generated anion species with an electrophile was utilized. Recently, Yoshida and co-workers reported on a three component reaction of benzyne, amine, and carbon dioxide to provide anthranilic acid.6g; (b) T. M. Sielecki and A. I. Meyers, J. Org. Chem., 1992, 57, 3673 CrossRef CAS; (c) H. Yoshida, E. Shirakawa, Y. Honda and T. Hiyama, Angew. Chem., Int. Ed., 2002, 41, 3247 CrossRef CAS; (d) H. Yoshida, T. Minabe, J. Ohshita and A. Kunai, Chem. Commun., 2005, 42, 3454 RSC; (e) E. Yoshioka, S. Kohtani and H. Miyabe, Angew. Chem., Int. Ed., 2011, 50, 6638 CrossRef CAS; (f) H. Yoshida, Y. Ito and J. Ohshita, Chem. Commun., 2011, 47, 8512 RSC; (g) H. Yoshida, T. Moroshita and J. Ohshita, Org. Lett., 2008, 10, 3845 CrossRef CAS; (h) Recently, Garg and co-workers reported nucleophilic addition to indolyne and its application to synthesis of indolactam V: S. M. Bronner, A. E. Goetz and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 3832 CrossRef CAS.
- (a) K. Okano, H. Fujiwara, T. Noji, T. Fukuyama and H. Tokuyama, Angew. Chem., Int. Ed., 2010, 49, 5925 CAS; (b) H. Tokuyama, K. Okano, H. Fujiwara, T. Noji and T. Fukuyama, Chem.–Asian J., 2011, 6, 560 CrossRef CAS.
- J. H. Tidwell and S. L. Buchwald, J. Org. Chem., 1992, 57, 6380 CrossRef CAS . The overall yield was improved by the optimization of each step.
- (a) Tricyclic compound 11 was obtained in low to moderate yields with the following bases: Mg(TMP)2·2LiBr,9b Mg(TMP)2·2LiCl,9c LiAl(TMP)(i-Bu)3,9d Li(TMP)ZnMe2,9e,9f and MeCu(TMP)(CN)Li29g; (b) P. E. Eaton, C.-H. Lee and Y. Xiong, J. Am. Chem. Soc., 1989, 111, 8016 CrossRef CAS; (c) G. C. Clososki, C. J. Rohbogner and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 7681 CrossRef CAS; (d) M. Uchiyama, H. Naka, Y. Matsumoto and T. Ohwada, J. Am. Chem. Soc., 2004, 126, 10526 CrossRef CAS; (e) D. V. Graham, E. Hevia, A. R. Kennedy and R. E. Mulvey, Organometallics, 2006, 25, 3297 CrossRef CAS; (f) M. Uchiyama, Y. Kobayashi, T. Furuyama, S. Nakamura, Y. Kajihara, T. Miyoshi, T. Sakamoto, Y. Kondo and K. Morokuma, J. Am. Chem. Soc., 2008, 130, 472 CrossRef CAS; (g) S. Usui, Y. Hashimoto, J. V. Morey, A. E. H. Wheatley and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 15102 CrossRef CAS.
- R. A. Olofson and C. M. Dougherty, J. Am. Chem. Soc., 1973, 95, 582 CrossRef CAS.
- G. H. Hakimelahi and G. Just, Tetrahedron Lett., 1979, 20, 3643 CrossRef.
-
Procedure for benzyne-mediated cyclization-chlorination: A flame-dried 50-mL Schlenk tube equipped with a magnetic stirrer bar and an inlet adapter with a three-way stopcock was charged with 2,2,6,6-tetramethylpiperidine (1.70 mL, 10.0 mmol) and dry THF (20 mL) under an argon atmosphere. To the solution was added n-BuLi (1.60 M in n-hexane, 6.14 mL, 10.0 mmol) at −78 °C. The resulting solution was warmed to 0 °C over a period of 25 min. The resulting pale yellow solution was added to the solution of 10 (980 mg, 2.00 mmol) in THF (20 mL) at −78 °C dropwise over 5 min. The reaction mixture was stirred for 10 min, after which time TLC (hexanes/EtOAc = 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) indicated complete consumption of 10. To the reaction mixture was added 1,1,1,2,2,2-hexachloroethane (2.37 g, 10.0 mmol) at −78 °C. The resulting suspension was warmed to 0 °C for 30 min. The reaction mixture was treated with saturated aqueous ammonium chloride, and the mixture was extracted with EtOAc three times. The organic extracts were dried over anhydrous sodium sulfate, and filtered. The organic solvents were removed under reduced pressure to give a crude material, which was purified by column chromatography on silica gel to provide the title compound 12 (506 mg, 1.27 mmol, 64%) as a pale yellow amorphous solid; IR (neat, cm−1) 2979, 2936, 1704, 1608, 1487, 1450, 1409, 1378, 1330, 1303, 1169, 1138, 760; 1H NMR (400 MHz, CDCl3) δ 7.40–7.25 (m, 0.8H), 6.87–7.06 (m, 0.2H), 4.38 (t, 1H, J = 1.0 Hz), 4.18–4.35 (m, 2H), 3.92–4.07 (m, 1H), 3.91 (s, 3H), 3.60 (dd, 1H, J = 11.2, 8.4 Hz), 3.45–3.28 (m, 1H), 3.26–3.14 (m, 1H), 2.54–2.40 (m, 1H), 1.56–1.44 (m, 1H), 1.49 (s, 9H), 1.44–1.26 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 156.3, 153.5, 152.9, 139.6, 134.5, 121.2, 111.3, 97.0, 81.2, 61.4, 56.7, 55.4, 44.3, 32.8, 31.2, 27.9, 14.5; HRMS (ESI+) cald. for C19H25ClN2O5 (M+), 396.1452; found 396.1436.
:1) indicated complete consumption of 10. To the reaction mixture was added 1,1,1,2,2,2-hexachloroethane (2.37 g, 10.0 mmol) at −78 °C. The resulting suspension was warmed to 0 °C for 30 min. The reaction mixture was treated with saturated aqueous ammonium chloride, and the mixture was extracted with EtOAc three times. The organic extracts were dried over anhydrous sodium sulfate, and filtered. The organic solvents were removed under reduced pressure to give a crude material, which was purified by column chromatography on silica gel to provide the title compound 12 (506 mg, 1.27 mmol, 64%) as a pale yellow amorphous solid; IR (neat, cm−1) 2979, 2936, 1704, 1608, 1487, 1450, 1409, 1378, 1330, 1303, 1169, 1138, 760; 1H NMR (400 MHz, CDCl3) δ 7.40–7.25 (m, 0.8H), 6.87–7.06 (m, 0.2H), 4.38 (t, 1H, J = 1.0 Hz), 4.18–4.35 (m, 2H), 3.92–4.07 (m, 1H), 3.91 (s, 3H), 3.60 (dd, 1H, J = 11.2, 8.4 Hz), 3.45–3.28 (m, 1H), 3.26–3.14 (m, 1H), 2.54–2.40 (m, 1H), 1.56–1.44 (m, 1H), 1.49 (s, 9H), 1.44–1.26 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 156.3, 153.5, 152.9, 139.6, 134.5, 121.2, 111.3, 97.0, 81.2, 61.4, 56.7, 55.4, 44.3, 32.8, 31.2, 27.9, 14.5; HRMS (ESI+) cald. for C19H25ClN2O5 (M+), 396.1452; found 396.1436.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H and 13C NMR spectra. See DOI: 10.1039/c2ra20604h |
| This journal is © The Royal Society of Chemistry 2012 |