Specific uptake and interactions of peptide nucleic acid derivatives with biomimetic membranes†
Tanmaya
Joshi
a,
Gilles
Gasser
*c,
Lisandra L.
Martin
*a and
Leone
Spiccia
*ab
aSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: Leone.Spiccia@monash.edu; Lisa.Martin@monash.edu; Fax: +61 3 9905 4597
bARC Centre of Excellence for Electromaterials Science, Monash University, Clayton, Victoria 3800, Australia
cInstitute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland. E-mail: gilles.gasser@aci.uzh.ch; Fax: +41 44 635 68 03
First published on 15th March 2012
Abstract
There is a growing interest in understanding the uptake mechanism of metal-containing peptide nucleic acid (PNA) bioconjugates into living cells. In this study, quartz crystal microbalance with dissipation monitoring (QCM-D) has been used to explore the membrane specific uptake and interactions of PNA/peptide/Ru(II) conjugates. For all lipid compositions, the unmodified PNA oligomer and its Ru(II) conjugate were found to traverse freely across the membrane in a trans-membrane manner, causing no significant changes in the membrane structure. The nuclear localised signal peptide (NLS) conjugated sequences showed membrane specific activities. In model mammalian and bacterial-mimetic membranes, rapid trans-membrane insertion was observed followed by a concentration dependent disruption and irreversible structural changes to the membrane system. The variations in the magnitude of the structural changes and in their tendency to facilitate disruption are ascribed to hydrophobicity, the cationic charge introduced on modification of the original PNA backbone as well as the physical state of the model membrane used.
Introduction
Peptide nucleic acids (PNAs)1 are considered promising candidates for gene therapeutics.2 However, poor cellular uptake and limited in vivo bioavailability have, to date, restricted their application in this field.3–9 Despite possessing a neutral pseudopeptide backbone, PNAs are large and hydrophilic molecules. This implies that cellular administration in an unmodified form cannot be readily achieved.10–11 Several methods of delivery as well as modifications to the PNA oligomers have been suggested for improving their cellular uptake. These include co-delivery with partial cDNA oligonucleotides, cationic lipids and lipophilic triphenylphosphonium (TPP) cations, and a few examples of chemical modifications of PNA oligomers with metal complexes.6,8,11–15 However, to date, the most promising approach has been the conjugation of PNAs with cell penetrating peptides (CPP).6,8,12 Although the mechanism of CPP translocation is not fully understood, their ability to successfully transport cargo (e.g., PNAs) into the cell makes them effective delivery agents.12,16–18 However, this does not necessarily correlate with an improvement in PNA antisense activity. The issue is that internalization occurs predominantly via an endocytotic pathway, leading to the PNAs being delivered into an endosomal compartment and thereafter being trapped inside the cell.5,6,8,17In order to enhance the antisense effects of PNAs, better PNA cellular delivery methods are required as well as the elucidation of the factors influencing endosomal escape. Overcoming the poor distribution of PNAs in either the cytoplasm or nucleus, if achieved, could enhance their bioactivity.4,6,8,13,17,19,20 Although recent approaches applying endosomolytic agents, such as chloroquine and Ca2+, photochemical internalization, conjugation with a cationic peptide-lipid (CatLip) domain or nuclear localisation signal (NLS) peptides6,11,15,17,21,22 have shown promising improvements in cellular bioavailability, there is still a need to improve our understanding of cellular uptake for different delivery vectors and to provide better correlations of their in vivo structure–activity relationships.
Research in our group has focused on biophysical studies of peptide–membrane interactions using quartz crystal microbalance with dissipation monitoring (QCM-D).23–28 QCM-D is a highly sensitive, convenient tool for the quantitative and qualitative analysis of biomolecular interactions.29,30 It allows for real time monitoring of mass and structural changes in the lipid membrane upon their interaction with biomolecules providing unique fingerprints for each peptide.26,29–33 In this study, QCM-D has been used to study the interaction of a short PNA oligomer with a covalently linked NLS peptide (PKKKRKV)21,34,35 and/or Ru(II)-polypyridyl complexes in bacterial and mammalian-mimetic membranes. Although the precise mechanism for cellular delivery of PNA–peptide–metal bioconjugates remains unclear, these QCM-D studies using model bacterial and mammalian membranes are expected to reasonably reflect the major interactions occurring between the modified PNA–peptide–metal bioconjugates and the lipid bilayer, hence, mimicking the processes at the cellular level.23,24,36
In general terms, this article represents a significant addition to the growing field of research focusing on metal-containing PNAs.13 Although much attention has been focused on the preparation of such constructs,37–41 only recently have papers appeared that report the applications of non-radioactive metal bioconjugates in living cells.14–15,42–44 It is therefore important to understand the mechanism of uptake of these nucleotide analogues.
Experimental
Chemicals
Sodium chloride, potassium phosphate monobasic and potassium phosphate dibasic, cholesterol, chloroform (≥ 99.8%) and methanol (≥ 99.9%) were purchased from Sigma-Aldrich. The synthetic phospholipids derivatives, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol) (sodium salt) (DMPG), were purchased from Avanti Polar Lipids (Alabaster, USA). 3-Mercaptopropionic acid (MPA) was purchased from Fluka, BioChimica (Buchs, Switzerland). Ultrapure water with an initial resistivity of 18.2 MΩ cm was used for all experiments. Phosphate buffered saline (20 mM KH2PO4 and K2HPO4, pH 6.9 ± 0.1) with 100 mM sodium chloride (high-salt PBS) and 10 mM sodium chloride (low-salt PBS) were prepared in water.Synthesis of PNA oligomers
The synthesis and characterization of the oligomeric sequences used in this study, namely PNA1 (H–g-c-a-a-t-a-a-a-a-lys-NH2), PNA2 (H-g-c-a-a-t-a-a-a-a-M1-lys-NH2), PNA3 (H-PKKKRKV-g-c-a-a-t-a-a-a-a-lys-NH2) and PNA4 (H-PKKKRKV-g-c-a-a-t-a-a-a-a-M1-lys-NH2) as well as M1 = [Ru(bpy)2(Cpp–L–PNA–OH)](PF6)2 have been previously described.45 All sequences were prepared by manual solid phase synthesis according to standard protocols.43,44,46,47 PNA sequences are written from the N- to C- terminus with the small letters a, t, g and c representing PNA monomers. PNA stock solutions were prepared in ultrapure water and stored at −18 °C. The PNA strand concentrations of the stock solutions were determined by UV spectroscopy45 and used to prepare the desired concentrations of PNA oligomers in high-salt PBS.Liposome preparation
5 mM stock solutions of DMPC, DMPG and cholesterol, prepared by individually dissolving DMPC and cholesterol in chloroform and DMPG in chloroform/methanol (ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), were used in appropriate volume ratios needed for obtaining the desired lipid compositions (DMPC, DMPC/cholesterol 7:3 v/v, DMPC/DMPG 4:1 v/v). The lipid solutions were dried under N2, followed by vacuum desiccation. Liposomes were prepared by re-suspending the lipids in high-salt PBS at 0.5 mM concentration and incubation at 37 °C, followed by vortexing and brief sonication (ca. 5 min) prior to use. The resultant liposomes are unilamellar with bimodal size distribution (average radii for the two populations ca. 50 and 300 nm).36
:1 v/v), were used in appropriate volume ratios needed for obtaining the desired lipid compositions (DMPC, DMPC/cholesterol 7:3 v/v, DMPC/DMPG 4:1 v/v). The lipid solutions were dried under N2, followed by vacuum desiccation. Liposomes were prepared by re-suspending the lipids in high-salt PBS at 0.5 mM concentration and incubation at 37 °C, followed by vortexing and brief sonication (ca. 5 min) prior to use. The resultant liposomes are unilamellar with bimodal size distribution (average radii for the two populations ca. 50 and 300 nm).36
Quartz crystal microbalance with dissipation monitoring (QCM-D)
The QCM-D measurements were performed using an E4 system with flow cells (Q-Sense, Västra Frölunda, Sweden). The polished, gold-coated, AT-cut quartz chips with a fundamental oscillating frequency of ca. 5 MHz, were used as sensor crystals. All experiments were conducted 19.10 ± 0.05 °C and in triplicate to ensure reproducibility of the results. In the QCM experiments, the relative changes in resonance frequency (Δf) and energy dissipation (ΔD) of the chip were simultaneously recorded at the 1st, 3rd, 5th, 7th and 9th harmonics. The raw data was obtained as ASCII files and analysed with OriginPro 8 (OriginLab, Northampton, USA). Data for the 1st harmonic (i.e., fundamental frequency of the crystal) was not included in the analysis as it is generally unreliable.25,26 Unless otherwise stated, only the 7th harmonic data plots are presented for discussion.Before each experiment, the surface of the chips was chemically modified following a reported protocol.26 Specifically, the chips were immersed in a NH4OH:H2O2:H2O (1:1:3 v/v) mixture for 20–25 min at ca. 70 °C and rinsed thoroughly with water afterwards. The freshly cleaned chips were immersed in 1 mM MPA in propan-2-ol for 1 h to create an MPA based anionic self-assembled monolayer on the gold surface. Any excess of physically adsorbed MPA was removed by rinsing thoroughly with propan-2-ol and drying under N2 prior to use.
QCM-D experiments were devised based on published protocol.26 In a typical QCM experiment, liposome solution was flushed into the QCM-D chamber (50 μL min−1) for their adsorption onto the MPA monolayer, which is followed by the process of deformation, rupturing and fusion, leading to the formation of lipid bilayer on the crystal surface.36 The chamber was sequentially flushed with high salt and low salt PBS (300 μL min−1) to ensure removal of any weakly bound liposomes as well as facilitate rupturing of any intact lipid vesicles, as a result of the difference in osmotic pressure. A 1 mL aliquot of a PNA solution of known concentration was then introduced (flow rate 50 μL min−1) in the chamber and left to incubate with the lipid membrane. A final high salt PBS wash (300 μL min−1) was then carried out to aid the assessment of the reversibility of any process that may be occurring. The mass deposition on the crystal was calculated from the change in frequency for the 7th harmonic (unless otherwise stated) and use of the Sauerbrey equation.26,33,48
Results and discussion
Membrane binding of PNA sequences
The QCM-D technique was used to probe the interactions between the PNA sequences (see Table 1 and Fig. 1) and the phospholipid bilayers. An unmodified PNA oligomer (PNA1) and the Ru(II)-polypyridyl and/or NLS peptide based conjugates (PNA2, PNA3 and PNA4) have been studied to allow comparison of their respective membrane responses and to probe the effect of these modifications on the inherent plasma membrane affinity of PNAs.49–52 Since factors such as physiological state of the target cells and cell surface components also influence the uptake mechanism, PNA–lipid interactions were studied on zwitterionic (neat DMPC (PC) and DMPC/cholesterol (PC-Chol)) and anionic (DMPC/DMPG (4:1) (PCPG)) lipid compositions that bear considerable resemblance to the mammalian and bacterial membrane surfaces in a protein free environment.53,54 These studies were carried out using 1, 2, 5 and 10 μM PNA solutions.
 | ||
| Fig. 1 Structure of the Ru(II)–PNA-like monomer, M1. | ||
| PNA code | Sequencea | Chargec |
|---|---|---|
| a Small letters a, t, g, c denote PNA monomers and the amino acids are represented using standard one letter codes. PNA sequences are written from N- to C- terminus. b See Fig. 1 for the structure of M1. c pH 6.9 ± 0.1. | ||
| PNA1 | H-g-c-a-a-t-a-a-a-a-lys-NH2 | + 2 |
| PNA2 | H-g-c-a-a-t-a-a-a-a-M1-lys-NH2b | + 4 |
| PNA3 | H-PKKKRKV-g-c-a-a-t-a-a-a-a-lys-NH2 | + 7 |
| PNA4 | H-PKKKRKV-g-c-a-a-t-a-a-a-a-M1-lys-NH2b | + 9 |
The PNA–lipid membrane interactions were analyzed using the time-dependent frequency changes (Δf-t), time-dependent energy dissipation changes (ΔD-t) and energy dissipation vs. frequency changes (Δf-ΔD) plots.23–28 Changes in the oscillation frequency of chip (Δf) in QCM-D gives information about the mass deposited on the surface, according to the Sauerbrey relationship. For a tightly coupled material, the change in frequency (−Δf) is directly correlated with mass (Δm), where Δm corresponds to the mass attached to the sensor surface.48 In addition, the viscoelastic properties of the chip surface can be measured using the change in energy dissipation, ΔD. Increasing energy dissipation (ΔD > 0) provides evidence for the adsorbed film on the chip becoming thicker, softer or looser. On the other hand, a decrease in change of energy dissipation suggests the transformation to a more rigid and compact structure.30,33,55
The QCM-D technique can also determine the penetration depth for a particular frequency wave, holding an inverse relationship with the overtone number.56 This implies that the higher harmonics can probe more closely to the sensor surface and vice versa. Thus, a comprehensive understanding including the kinetics of PNA–membrane interactions in a three dimensional membrane structure can be readily obtained by measurements at several different harmonics.23 For this QCM-D study, the change in frequency (Δf) and energy dissipation (ΔD) of the sensor chip were measured at the first, third, fifth, seventh and ninth harmonics, although the data from only 3rd–9th was used for analysis. Furthermore, these data gave rise to the Δf-ΔD plots which provided the fingerprints for interaction at the membrane coated chip surface. These plots have previously proven complementary in terms of exposing mass and structural changes occurring due to interaction between the phospholipid bilayer and the biomolecules introduced at the membrane–water interface.24–28
DMPC/cholesterol (PC-Chol) based phospholipid bilayer
Introduction of PNAs into the QCM-D resulted in immediate binding onto the membrane, as shown in the Δf-t plot for PNA1 (Fig. 2 (top)), and in a mass gain with time until a threshold limit was obtained. As is apparent from the Δf-t curve, the mass addition was concentration dependent, with a greater mass uptake and more rapidly attained threshold level at the highest PNA1 concentration (10 μM).The diffusion of the unmodified PNA (PNA1) into the membrane appears to be energetically favorable, as evident from the mass uptake even at low PNA concentrations. The frequency change, measured for the different harmonics (vide supra), was independent of the harmonic number (representative data shown in Fig. S1, ESI†). These data suggest a uniform trans-membrane mass density across the membrane. This effect was observed at all PNA1 concentrations, irrespective of the variable mass uptake. The membrane uptake of the PNA was also quite reversible with almost immediate and complete removal of PNA1 from the membrane with the final high salt PBS wash (Fig. 2). At the higher PNA1 concentrations (5 and 10 μM), however, the frequency did not return to the initial value following the buffer wash. This suggests that some residual PNA1 remains within the membrane (Δf ≈ −2 Hz at 10 μM). These data are combined with the change in dissipation, and represented using the Δf-ΔD fingerprint plots (Fig. 2 (bottom)) for PNA1 addition on the PC-Chol membrane. Interestingly, since ΔD is invariant over the concentration range studied, the insertion (and removal) of PNA1 from the lipid membrane is not accompanied by significant changes in the structural properties of the membrane.
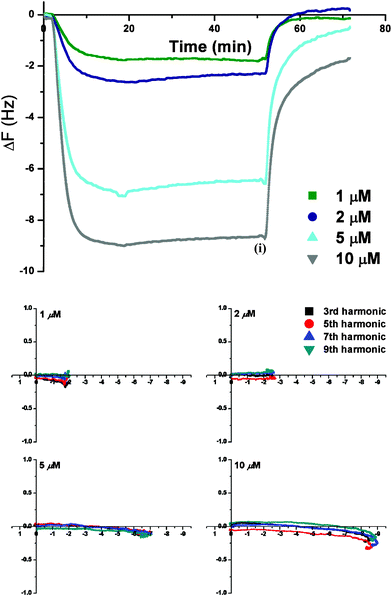 | ||
| Fig. 2 QCM-D monitoring of PNA1 on DMPC/cholesterol (PC-Chol) membrane with: (top) Δf-t plot showing effect of PNA1 concentration (1–10 μM) on its membrane uptake {time (i) (i.e., t = 50 min) corresponds to the onset of the final high salt PBS wash}; (bottom) Energy dissipation (ΔD) vs. frequency (Δf) dependence for PNA1 interactions with the PC-Chol membrane {The x and y-axis represent Δf and ΔD (10−6) values, respectively. The final buffer rinse (t ≥ 50 min) is omitted}. | ||
The physical interpretation using the Δf-t plot for multiple harmonics and the Δf-ΔD plots is consistent with non-disruptive trans-membrane23 insertion of the PNA1, into the membrane following a wash step with high salt PBS buffer solution. This process appears to be accompanied by equally favorable kinetics for the removal of PNA1 from the membrane. Consequently, there must be a rapid equilibrium established between oligomers partitioned in the aqueous phase and membrane. In contrast, Wittung and coworkers reported a slow efflux of fluorescein-labelled 10-mer PNA sequences from liposomes (efflux half time = 7 days), and concluded that the entry of PNA into living cells by passive diffusion through membrane is a slow process.10 In the same study, it was also suggested that lipophilic PNAs can be trapped in the aqueous compartments of the liposomes rather than being distributed in the lipid membrane.10 Molecular dynamics simulations for a 6-mer PNA sequence at a POPC bilayer in water by Weroński et al. showed that these sequences adsorbed at the lipid–water interface.57 This interfacial adsorption process was proposed to be driven by either the electrostatic attractions or the hydrophobic interactions, depending on the polarity of the terminal groups in PNA; the strength of the interaction as well as kinetics of the adsorption being driving force dependent.57 From simulations of lipid bilayer system under physiological conditions, it was further predicted that PNA adsorption at the interface could be prohibited due to strong adsorption of ions taking place at the lipid–water interface. The trans-membrane uptake of PNA1, as observed in the current study, differs from these earlier studies and demonstrates that simple PNA molecules can freely traverse across the cell membrane. However, this behavior has been observed for the antimicrobial peptides, apidaecin 1a and 1b and oncocin, in which the peptide addition and its almost complete removal from membrane occurred in a non-lytic manner.24,28
The QCM-D data for PNA2, an oligomeric sequence with a Ru(II)-polypyridyl complex attached at its C-terminus via an alkyl linker (Fig. 1), showed a response analogous to PNA1 (Fig. 3 (top)). The mass addition (Δf < 0) on the lipid bilayer attained a steady state with time, and was governed by the concentration of the PNA2. Overall, higher concentrations yielded a faster and greater PNA2 uptake. The increase in mass was uniform across the membrane bilayer, as deduced from the similar frequency change measured for all harmonics (Fig. S2† shows representative data at 5 μM).
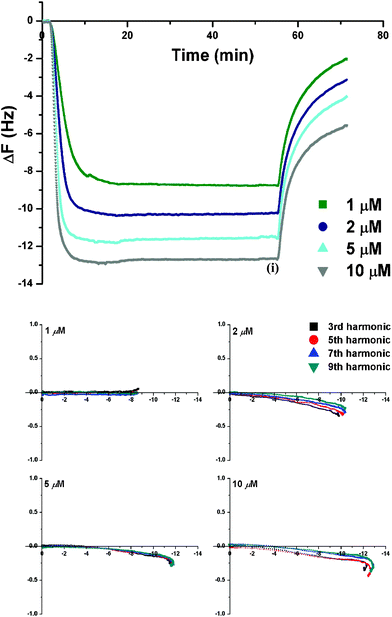 | ||
| Fig. 3 QCM-D monitoring of PNA2 on the DMPC/cholesterol (PC-Chol) membrane with: (top) Δf-t plot showing effect of PNA2 concentration (1–10 μM) on its membrane uptake {time (i) (i.e., t = 50 min) corresponds to the onset of the final high salt PBS wash}; (bottom) Energy dissipation (ΔD) vs. frequency (Δf) dependence for PNA2 interactions with PC-Chol membrane {The x and y-axis represent Δf and ΔD (10−6) values, respectively. The final buffer rinse (t ≥ 50 min) is omitted}. | ||
A significant amount of PNA2 was removed from the membrane with the final buffer wash (vide infra) indicating that the interaction of the PNA2 with the membrane is dependent on the concentration of PNA2 in the bulk solution. Interestingly, the same amount of PNA was removed during the high salt PBS wash (Δf = + (7.0 ± 0.3) Hz) irrespective of the PNA concentration used in the insertion process. The final mass retained in the membrane after the buffer wash ranged from 2.1–5.6 Hz across the concentration range (2–10 μM) investigated.
The Δf-ΔD curve (Fig. 3(bottom)) shows that for PNA2 the interaction with the membrane is not completely reversible, viz., PNA2 addition causes a small decrease in energy dissipation that does not return to its original value following the buffer wash. Binding of PNA2 to the membrane appears to cause a small but permanent change in the membrane structure. However, as found for PNA1, overlapping harmonics support a trans-membrane orientation for the inserted PNA2. Interestingly, the amount of PNA inserted and retained is higher for PNA2, vs.PNA1, for all the concentrations studied. This is indicative of stronger membrane association for PNA2.
The time dependent frequency plots for PNA3 and PNA4, the respective NLS peptide (PKKKRKV)34 conjugates of PNA1 and PNA2 (Table 1), differ significantly from the traces characteristic of their parent analogues (Fig. 4). The initial decrease in frequency implies rapid uptake of PNA3 and PNA4 into the membrane. PNA3 shows a concentration dependent rate of membrane insertion, however, a similar mass uptake was observed irrespective of the PNA concentration used (Δf ≈ −8.5 Hz). On the other hand, PNA4 showed a discontinuous trend in uptake with low PNA4 concentrations (1 and 2 μM) resulting in greater uptake (Δf ≈ −11.0 Hz). For the 2–10 μM PNA4 solutions the insertion rate was similar, but the threshold uptake limit decreased in the order 2 μM > 10 μM > 5 μM. Once the membrane was saturated, further addition of the PNA3 solution resulted in mass removal (increase in frequency). Interestingly, the threshold limit for PNA4 insertion into the membrane was higher at 2 μM concentration. Any further flow of solution into the chamber did not result in any mass removal for PNA4.
 | ||
| Fig. 4 Δf-t plot showing: (top) effect of PNA3 concentration (1–10 μM) on its interaction with PC-Chol membrane; (bottom) Concentration dependence of PNA4 interactions with PC-Chol lipid membrane. Time (i) and (ii) (i.e., t = 50 min) in respective graphs correspond to the onset of the final high salt PBS wash. | ||
For all concentrations, the frequency increment (during the mass removal process for PNA3 and PNA4) was highest for the 3rd harmonic (Fig. 5), with a decrease in magnitude observed with increasing overtone number (see Fig. S3–S4 for Δf-t plots for overtone effects at 10 μM PNA3 and PNA4). The overlapping harmonics for the initially decreasing frequency response suggests that PNA3 and PNA4 both inserted into the lipid membrane in a trans-membrane orientation until saturation was achieved. Material (PNA and lipid) removal from the membrane surface occurs following further addition of PNA3.
 | ||
| Fig. 5 Energy dissipation (ΔD) vs. frequency (Δf) dependence of PNA3 (top) and PNA4 (bottom) activity on PC-Chol membrane. The x and y-axis represent Δf and ΔD (10−6) values, respectively. The final buffer rinse is not shown. | ||
The rate of membrane association for PNA3 and PNA4 was compared with their parent analogues, PNA1 and PNA2, by plotting the first order derivative of the change in frequency (Δf) vs. time, ∂Δf/∂t. The plot revealed an interesting rate profile for mass addition/removal process. As can be gleaned from Fig. 6, rate of the PNA binding process follows the order, PNA4 > PNA2 > PNA3 > PNA1. Based on the trend exhibited by individual derivative curves, it can be said that whereas the rate of insertion into the membrane appears to be quite slow for PNA1, it is about 8-fold faster in case of PNA4. The derivative curve also provides strong qualitative evidence that while a NLS sequence conjugated to the PNA oligomer assists in increasing the rate of trans-membrane insertion (PNA3vs.PNA1), overall mass uptake into the PC-Chol membrane occurs at a faster rate for the Ru(II) based PNA sequences. Interestingly, the kinetic profile for material removal from the bilayer membrane during the final buffer wash showed no significant differences for the different PNA sequences. This suggests that the PNA concentration in the bulk solution is the primary parameter that controls the material removal process during the final buffer wash, irrespective of the nature of PNA sequence.
 | ||
| Fig. 6 First order derivative of 7th harmonic Δf-t traces, obtained for interaction of 10 μM PNA1–PNA4 with PC-Chol membrane, plotted against time. | ||
The Δf-ΔD plot (Fig. 5 (top)) was used to further understand how the PNA3 interacts with the lipid membrane. Firstly, Fig. 5 reconfirms the trans-membrane insertion of PNA3 and PNA4 with no structural change in the membrane (ΔD ≈ 0). However, further addition of PNA3 results in membrane saturation and the onset of material removal is accompanied by a decrease in energy dissipation (i.e ΔD < 0). A slightly smaller decrease in dissipation and greater mass removal was measured from ΔD and Δf readouts for the lower numbered harmonics. These data suggest that once PNA uptake by the membrane reaches a threshold limit, further addition of PNA3 causes a decrease in mass attributable predominantly to the removal of surface bound/partially inserted PNA. This phenomenon is consistent for all concentrations (1–10 μM) of PNA3, with the threshold always reached prior to the incubation stage (t < 20 min, refer to the Experimental section), indicating that further addition of PNA3 is necessary for mass removal and for structural reorganization to occur. This interpretation is supported by the fact that steady energy dissipation and frequency levels are maintained for PNA3 during the incubation stage and that further mass removal is the same for all harmonics (Δf = + (4.4 ± 0.1) Hz at 5 μM), with no change in energy dissipation during the final high salt PBS buffer wash. Somewhat similar membrane activity has previously been observed for the antimicrobial peptides, Caerin and Maculatin.23 The slow mass removal observed at higher concentrations (10–20 μM) for these peptides was proposed to be due to mixed micelle formation inciting material removal once a threshold concentration within membrane was reached. The fact that mass loss is independent of harmonics, as found for PNA1 and PNA2 (vide supra), suggests that this process is equilibrium driven and occurs homogenously across the membrane.
The interactive binding of PNAs with charged lipid membranes (prokaryotic mimics) is considered to involve contributions from electrostatic attractions, hydrophobic interactions and dehydration effects.58 Both PNA3 and PNA4 differ from their parent analogues in their hydrophobic regions and greater overall positive charge. Since the PC-Chol membrane model is zwitterionic, and experiments on PNA sequences (PNA1-PNA4) were carried out under similar ionic strength and pH conditions, hydrophobic contributions can be proposed to represent the major differentiating factor in their activity towards the PC-Chol membrane. The addition of a NLS peptide sequence (PKKKRKV) at the N-terminus and the Ru(II)-polypyridyl unit at the C-terminus in PNA4 increases the number of hydrophobic sites (from proline, valine and arginine residues, and an alkyl chain linked polypyridyl ligand) in the modified PNA sequences. Thus, we propose that these sites contribute to the higher membrane uptake potency of PNA3 and PNA4, as displayed from the saturation limit being attained quickly even at 1 μM concentration. During the trans-membrane insertion of PNA3, penetration due to the hydrophobic interactions between the phospholipid membrane and hydrophobic regions on the PNA sequences can be speculated. However, the QCM-D data indicate that a significant structural impact on the membrane can be forced with further PNA3 addition. The PNA molecule may be residing inside the membrane in a thermodynamically unstable state, because of the charged polylysine and arginine residues being located within the energetically unfavourable, non-polar environment inside the lipid membrane. As a result, further PNA addition drives structural reorganization inside the membrane surface, as indicated by a decrease in energy dissipation factor, which reflects the structural changes on the chip. For PNA4, the polypyridyl unit and alkyl linker are suggested to further increase the fraction of hydrophobic regions at its C-terminus. QCM-D data thus provides valuable insights into the chaperonic role of the attached NLS peptide and Ru(II)-polypyridyl unit in promoting PNA insertion into membrane. It should be noted, however, that aggregation of PNA on the membrane surface on further addition of the sample beyond the membrane threshold levels and subsequent material removal cannot be completely ruled out at this stage. Alternative techniques are thus currently being explored to provide further basis to our postulated mechanism for membrane insertion of these PNA sequences.
DMPC/DMPG (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1) (PCPG) and DMPC (PC) based phospholipid bilayers
:1) (PCPG) and DMPC (PC) based phospholipid bilayers
A comparison of the QCM-D responses for PNA1 on PCPG with those on PC-Chol reveals no significant differences (Fig. S5–S6†). The Δf-t, ΔD-t and Δf-ΔD plots support the trans-membrane insertion of PNA1 and its subsequent removal during the final PBS wash. However, the slight increase in residual PNA remaining in the membrane after the final buffer wash, viz., some mass was retained at 2 μM PNA1 concentration, again shows the higher affinity of PNA1 for the PCPG lipid membrane.
In contrast, PNA2 displayed a different behavior on the PCPG membrane at the higher concentrations (5–10 μM) (Fig. 7 and 8). At 5 μM, as the PNA2 concentration reached saturation within the membrane, mass removal was observed from the chip surface (based on an interpretation from different overtones) (Fig. 7) although this effect is quite small. This process was also observed during the incubation stage when no PNA2 was flowing into the cell. A small decrease in ΔD was also observed. This indicated some structural changes could result from the attached cationic Ru(II) moiety imposing a slow reorganization of the PNA2 in the anionic lipid domains.
 | ||
| Fig. 7 Δf-t plot showing overtone effect for PNA2 (5 μM) uptake on a DMPC/DMPG (4:1) membrane. Time (i)–(ii) (i.e., 20–50 min) refers to the equilibration stage and time (t) ≥ 50 min corresponds to final PBS rinse. | ||
 | ||
| Fig. 8 Energy dissipation (ΔD) vs. frequency (Δf) dependence of the activity of PNA2 on the DMPC/DMPG (4:1) membrane. The final buffer rinse is not shown. | ||
A similar process was observed at 10 μM PNA2 with the only noticeable difference being an increase in dissipation during the initial insertion process (Fig. 8). This could be due to increased electrostatic interactions between the anionic lipid head groups and the positively charged Ru(II)-PNA conjugate, PNA2. However, these two processes were only observed at the higher concentrations and QCM-D data alone is insufficient to establish the exact mechanism.
For PNA3 and PNA4, saturation of the membrane by insertion into the PCPG membrane was concentration dependent (Δf-t plots showing overtone effects for 10 μM PNA3 and PNA4 are presented in Fig. 9). For PNA3, insertion was rapid and was followed by membrane disruption even at low PNA3 concentrations. The larger frequency change measured for 3rd and 5th harmonics indicated that more mass was being lost from the upper leaflet of the membrane. The loss of mass was accompanied by a harmonic dependent decrease in energy dissipation, consistent with membrane removal. Thus, a decrease in membrane thickness would be observed as disruption mainly occurs from the membrane surface, as mentioned earlier. PNA4 demonstrated a similar interaction profile with the PCPG membrane as described for PC-Chol. The Δf-t traces for PNA4 showed that a slow mass removal begins at 5 μM concentration, with the removal being more pronounced at a 10 μM concentration. The Δf-ΔD plots (Fig. S7–S8†) reinforce these observations with saturation of a low dissipation binding of PNA4 preceding the removal of membrane–PNA material from the sensor. For both PNA3 and PNA4, the final PBS wash once again resulted in an additional mass loss with no further energy dissipation change (data not shown). A similar membrane activity has been discussed for the cell penetrating Tat (49–57) peptide on DMPC/DMPG 4:1 lipid composition in our earlier work.26
 | ||
| Fig. 9 Δf-t plot showing the overtone effect for (top) 10 μM PNA3 and (bottom) 10 μM PNA4 uptake on DMPC/DMPG (4:1) membrane. Time (t) ≥ 50 min corresponds to final PBS rinse. | ||
The rate of PNA binding (first order derivative of the Δf-t traces, ∂Δf/∂t) for 10 μM PNA1–PNA4 plotted against time (Fig. 10) showed the rate of mass addition to increase in the order PNA1 < PNA2 < PNA4 ≤ PNA3. The respective rate profiles for mass uptake into the anionic PCPG membrane present a direct correlation with the overall formal charges on the PNA sequence (Table 1). The uptake into the membrane for NLS peptide bearing PNA3 and PNA4 appears to be up to two folds faster than the Ru(II)-PNA bioconjugate, PNA2 (Fig. 10). The rate profile with mass increment yet again highlights that the process of addition is slowest for the unmodified PNA1. Subsequent attachment of cationic moieties in PNA2–PNA4 assists in improving the uptake of PNAs into the anionic membrane. Material removal observed during the final buffer wash displays almost identical rate profile for all PNAs. This further strengthens our previous speculation that the concentration of PNA in the bulk solution guides the process of removal of PNA from the membrane.
 | ||
| Fig. 10 The first order derivative of 7th harmonic Δf-t traces obtained for interaction of 10 μM PNA1–PNA4 with PCPG membrane, as plotted against time. | ||
The increased binding of the positively charged PNA3 and PNA4 sequences towards the PCPG membrane followed by greater membrane disruption, compared with PC-Chol membranes, highlights the important contribution of electrostatic attraction to the anionic membranes. It may be possible for the hydrophobic regions on the PNA sequence to facilitate insertion into the membrane; however, the propensity of lysine and arginine residues to structurally reorganize after insertion into the membrane, due to strong electrostatic interactions with the anionic head-groups from the DMPG component, may cause a greater destabilization leading to rapid membrane disruption. The situation is more complicated for PNA4. In this case, hydrophobic interactions and electrostatic attraction are both crucial for binding and penetration of the anionic surface, and a contribution from the flexible Ru(II)-polypyridyl unit at the distant C-terminus cannot be ignored. As anticipated, the stronger binding and less disruptive properties of PNA4, vs.PNA3, indicate a significant contribution from the Ru(II) unit towards its membrane activity. The hydrocarbon-linked Ru(II) unit may allow the inserted PNA4 sequence to reside in a more favorable configuration than PNA3. However, the involvement of other secondary factors including the difference in the length of the respective sequences cannot be completely ruled out.26
All experiments were performed at 19.10 ± 0.05 °C, below the phase transition temperature (Tm = 23 °C) of the DMPC lipid, implying that the membrane bilayers will be in the gel-phase.26 In the gel-phase, the membranes are rigid and have more defects, making them susceptible to disruption.26 Addition of cholesterol decreases the Tm of DMPC and changes the physical state of the membrane to a liquid crystalline phase, also known to reduce membrane stiffness.26,36,59 Studies have shown that the membrane activity of a particular molecule can be phase dependent.26 Thus, experiments on the neat DMPC (PC) lipid allowed us to comment on the phase-dependence of PNA1–PNA4, and the interaction with the lipid bilayer. The respective Δf-t plots for 5 μM PNA3 and PNA4 are shown in Fig. 11 with representative Δf-ΔD plots for 5 μM PNA1-PNA4 shown in Fig. S9–S12.†
 | ||
| Fig. 11 Δf-t plot showing the overtone effect for (top) PNA3 (5 μM) and (bottom) PNA4 (5 μM) uptake on a neat DMPC membrane. The final PBS rinse is not shown. | ||
PNA1 and PNA2 continued to exhibit the trend of trans-membrane insertion, with no changes in activity towards the PC membrane as compared to PC-Chol membrane. For PNA3 and PNA4, while the rapid trans-membrane insertion along with a decrease in energy dissipation did occur, a second process of mass removal was not observed. Instead, the overlapping harmonics were subsequently followed with their gradual separation and a further small decrease in Δf values for each harmonics, prior to them eventually attaining a steady state. The large −Δf values sensed for higher harmonics (9th > 7th > 5th > 3rd harmonic) in this process could correspond to an addition of mass into the membrane followed by its immediate redistribution from the top leaflet to the bottom leaflet or, if multi-layers are present, their penetration to the deeper bilayers, implying that the 7th and 9th harmonics will indicate greater mass gain. The first order derivative for the Δf-t traces vs. time graph (Fig. 12) showed that the rate of mass addition was greatest for PNA3, while being almost similar in the case of Ru(II)-PNA conjugates, PNA2 and PNA4. On the basis of the rate profile, it can also be concluded that mass uptake into the membrane is extremely slow for PNA1. This observation is in agreement with the poor membrane uptake that is generally observed for unmodified PNAs.
 | ||
| Fig. 12 The first order derivative of 7th harmonic Δf-t traces obtained for the interaction of 5 μM PNA1–PNA4 with neat DMPC membrane as plotted against time. The final PBS wash is not shown. | ||
As a universal control experiment, the PNA1–PNA4 solutions were also added to a mercaptopropionic acid (MPA) modified gold chip, in the absence of any lipid (data not shown). An addition of mass with sample flow, increasing in the order PNA1 < PNA2 < PNA3 < PNA4 was observed, along with no change in energy dissipation, ΔD ≈ 0. This reflects an electrostatic attraction between the negatively charged monolayer and cationic PNA sequences. Depending upon the overall positive charge carried by the PNA sequence (PNA4 > PNA3 > PNA2 > PNA1) the amount of PNA binding to the MPA monolayer changed, and the rate of mass gain decreased. This could be due to decreased electrostatic attraction as more and more PNA is bound to the monolayer. Thus, the observed activities for sequences PNA1–PNA4 are not simply an outcome of electrostatic binding to the anionic MPA monolayer but their inherent responses to different biomimetic phospholipid membranes.
Conclusions
We have investigated the uptake and interactions of an unmodified PNA oligomer (PNA1) and its Ru(II)-polypyridyl and/or NLS peptide based conjugates (PNA2, PNA3 and PNA4) with mammalian and bacterial mimetic membranes (DMPC/cholesterol and DMPC/DMPG (4:1), respectively). PNA1 and PNA2 showed a general tendency for trans-membrane insertion, at all lipid compositions. This insertion, as well as removal of the material proceeded with no significant perturbation of the membrane structure. A dynamic equilibrium is suggested to exist between PNA concentrations partitioned in the aqueous phase and membrane interior, as dissipation values were consistently low. The mass retained following the final buffer wash was greater for DMPC/DPMG than for DMPC/cholesterol membranes, with a residual mass of PNA2 > PNA1. The NLS/Ru(II)-polypyridyl conjugated sequences, PNA3 and PNA4, displayed stronger membrane binding affinity at all lipid compositions. In DMPC/cholesterol membranes, PNA3 and PNA4 showed rapid trans-membrane insertion followed by disruption of the membrane layers. The effect was magnified for the anionic phospholipids membrane (DMPC/DMPG (4:1)). Interestingly, PNA3 showed a greater disruptive activity compared to the PNA4. The magnitude of changes observed in the membrane structure, as well as variations in the tendency to facilitate disruption could be related to the increased number of hydrophobic sites and the greater cationic charge introduced through modifications on the original PNA backbone. Secondary factors, such as the relative length of sequences and physical state of the membranes, could also influence the PNA uptake and hence the membrane interactions. No disruption to the neat DMPC membranes was observed for PNA3 and PNA4, and this suggested that their activity could depend on the membrane phase.
Acknowledgements
This work was supported by the Australian Research Council through the Australian Centre of Excellence for Electromaterials Science (L.S.) and Discovery Grants (L.L.M.), the Swiss National Science Foundation (Ambizione Fellowship N° PZ00P2_126404 and Professorship N° PP00P2_133568 to G.G.) and the University of Zurich. T.J. is a recipient of a Monash Graduate Scholarship and a Monash International Postgraduate Research Scholarship. G.G. thanks Prof. Roger Alberto for generous access to all the facilities at the Institute of Inorganic Chemistry of the University of Zurich.References
- P. Nielsen, M. Egholm, R. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- P. E. Nielsen, ChemBioChem, 2010, 11, 2073 CrossRef CAS and references therein.
- P. E. Nielsen, Pharmacol. Toxicol., 2000, 86, 3–7 CAS.
- U. Koppelhus, S. K. Awasthi, V. Zachar, H. U. Holst, P. Ebbesen and P. E. Nielsen, Antisense Nucleic Acid Drug Dev., 2002, 12, 51–63 CrossRef CAS.
- U. Koppelhus and P. E. Nielsen, Adv. Drug Delivery Rev., 2003, 55, 267–280 CrossRef CAS.
- P. E. Nielsen, Q. Rev. Biophys., 2005, 38, 345–350 CrossRef CAS.
- K. E. Lundin, L. Good, R. Stroemberg, A. Graeslund and C. I. E. Smith, Adv. Genet., 2006, 56, 1–51 CrossRef CAS.
- P. E. Nielsen, Chem. Biodiversity, 2010, 7, 786–804 CAS.
- L. Good and P. E. Nielsen, Antisense Nucleic Acid Drug Dev., 1997, 7, 431–437 CrossRef CAS.
- P. Wittung, J. Kajanus, K. Edwards, P. Nielsen, B. Nordén and B. G. Malmström, FEBS Lett., 1995, 365, 27–29 CrossRef CAS.
- F. W. Rasmussen, N. Bendifallah, V. Zachar, T. Shiraishi, T. Fink, P. Ebbesen, P. E. Nielsen and U. Koppelhus, Oligonucleotides, 2006, 16, 43–57 CrossRef CAS.
- T. Shiraishi and P. E. Nielsen, in Delivery Technologies for Biopharmaceuticals, John Wiley & Sons, Ltd, 2009, pp. 305–338 Search PubMed.
- G. Gasser, A. M. Sosniak and N. Metzler-Nolte, Dalton Trans., 2011, 7061–7076 RSC.
- A. Füssl, A. Schleifenbaum, M. Göritz, A. Riddell, C. Schultz and R. Krämer, J. Am. Chem. Soc., 2006, 128, 5986–5987 CrossRef.
- S. I. Kirin, I. Ott, R. Gust, W. Mier, T. Weyhermüller and N. Metzler-Nolte, Angew. Chem. Int. Ed., 2008, 47, 955–959 CrossRef CAS.
- S. Veldhoen, S. Laufer and T. Restle, Int. J. Mol. Sci., 2008, 9, 1276–1320 CrossRef CAS.
- T. Shiraishi and P. E. Nielsen, Nat. Protocols, 2006, 1, 633–636 CAS.
- J. Howl, I. D. Nicholl and S. Jones, Biochem. Soc. Trans., 2007, 35, 767–769 CrossRef CAS.
- U. Soomets, M. Hallbrink and U. Langel, Front. Biosci., 1999, 4, D782–786 CrossRef CAS.
- K. W. Liang, F. Liu and L. Huang, Lett. Pept. Sci., 2004, 10, 161–167 CrossRef.
- K. Braun, P. Peschke, R. Pipkorn, S. Lampel, M. Wachsmuth, W. Waldeck, E. Friedrich and J. Debus, J. Mol. Biol., 2002, 318, 237–243 CrossRef CAS.
- T. Shiraishi and P. E. Nielsen, FEBS Lett., 2006, 580, 1451–1456 CrossRef CAS.
- A. Mechler, S. Praporski, K. Atmuri, M. Boland, F. Separovic and L. L. Martin, Biophys. J., 2007, 93, 3907–3916 CrossRef CAS.
- S. Piantavigna, P. Czihal, A. Mechler, M. Richter, R. Hoffmann and L. Martin, Int. J. Pept. Res. Ther., 2009, 15, 139–146 CrossRef CAS.
- G. McCubbin, S. Praporski, S. Piantavigna, D. Knappe, R. Hoffmann, J. Bowie, F. Separovic and L. Martin, Eur. Biophys. J., 2011, 40, 437–446 CrossRef CAS.
- S. Piantavigna, G. A. McCubbin, S. Boehnke, B. Graham, L. Spiccia and L. L. Martin, BBA - Biomembranes, 2011, 1808, 1811–1817 CrossRef CAS.
- P. J. Sherman, R. J. Jackway, J. D. Gehman, S. Praporski, G. A. McCubbin, A. Mechler, L. L. Martin, F. Separovic and J. H. Bowie, Biochemistry, 2009, 48, 11892–11901 CrossRef CAS.
- D. Knappe, S. Piantavigna, A. Hansen, A. Mechler, A. Binas, O. Nolte, L. L. Martin and R. Hoffmann, J. Med. Chem., 2010, 53, 5240–5247 CrossRef CAS.
- K. A. Marx, Biomacromolecules, 2003, 4, 1099–1120 CrossRef CAS.
- M. C. Dixon, J. Biomol. Tech., 2008, 19, 151–158 Search PubMed.
- C. A. Keller and B. Kasemo, Biophys. J., 1998, 75, 1397–1402 CrossRef CAS.
- N. J. Cho, C. W. Frank, B. Kasemo and F. Hook, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS.
- N.-J. Cho, K. K. Kanazawa, J. S. Glenn and C. W. Frank, Anal. Chem., 2007, 79, 7027–7035 CrossRef CAS.
- D. Kalderon, B. L. Roberts, W. D. Richardson and A. E. Smith, Cell, 1984, 39, 499–509 CrossRef CAS.
- G. Cutrona, E. M. Carpaneto, M. Ulivi, S. Roncella, O. Landt, M. Ferrarini and L. C. Boffa, Nat. Biotech., 2000, 18, 300–303 CrossRef CAS.
- A. Mechler, S. Praporski, S. Piantavigna, S. M. Heaton, K. N. Hall, M.-I. Aguilar and L. L. Martin, Biomaterials, 2009, 30, 682–689 CrossRef CAS.
- N. Metzler-Nolte, in Bioorganometallics: Biomolecules, Labeling, Medicine, ed. G. Jaouen, Wiley-VCH, Weinheim, 2006, pp. 125–179 Search PubMed.
- N. Metzler-Nolte, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer, Berlin Heidelberg, 2010, 32, 195–217 Search PubMed.
- C. Baldoli, P. Cerea, C. Giannini, E. Licandro, C. Rigamonti and S. Maiorana, Synlett., 2005, 13, 1984–1994 Search PubMed.
- G. Gasser, N. Hüsken, S. D. Köster and N. Metzler-Nolte, Chem. Comm., 2008, 3675–3677 RSC.
- A. Sosniak, G. Gasser and N. Metzler-Nolte, Org. Biomol. Chem., 2009, 7, 4992–5000 CAS.
- E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D'Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D'Alfonso and E. Licandro, Chem. Comm., 2010, 46, 6255–6257 RSC.
- G. Gasser, S. Neumann, I. Ott, M. Seitz, R. Heumann and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2011, 36, 5471–5478 CrossRef.
- G. Gasser, A. Pinto, S. Neumann, A. M. Sosniak, M. Seitz, K. Merz, R. Heumann and N. Metzler-Nolte, Dalton Trans., 2012, 41, 2304–2313 RSC.
- T. Joshi, G. J. Barbante, P. S. Francis, C. F. Hogan, A. M. Bond, G. Gasser and L. Spiccia, Inorg. Chem., 2012, 51, 3302–3315 CrossRef CAS.
- G. Gasser, K. Jäger, M. Zenker, R. Bergmann, J. Steinbach, H. Stephan and N. Metzler-Nolte, J. Inorg. Biochem., 2010, 104, 1133–1140 CrossRef CAS.
- N. Hüsken, G. Gasser, S. D. Koöster and N. Metzler-Nolte, Bioconjugate Chem., 2009, 20, 1578–1586 CrossRef.
- G. Sauerbrey, Zeitschrift für Physik, 1959, 155, 206–222 CrossRef CAS.
- L. Good and P. E. Nielsen, Nat Biotech, 1998, 16, 355–358 CrossRef CAS.
- L. Good and P. E. Nielsen, Proc. Natl. Acad. Sci. USA, 1998, 95, 2073–2076 CrossRef CAS.
- S. Sei, Q. E. Yang, D. O'Neill, K. Yoshimura, K. Nagashima and H. Mitsuya, J. Virol., 2000, 74, 4621–4633 CrossRef CAS.
- P. E. Nielsen, Expert Opin. Invest. Drugs, 2001, 10, 331–341 CrossRef CAS.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS.
- C. S. B. Chia, J. Torres, M. A. Cooper, I. T. Arkin and J. H. Bowie, FEBS Lett., 2002, 512, 47–51 CrossRef CAS.
- M. Rodahl, F. Hook, C. Fredriksson, C. A. Keller, A. Krozer, P. Brzezinski, M. Voinova and B. Kasemo, Faraday Discuss., 1997, 107, 229–246 RSC.
- M. Rodahl and B. Kasemo, Sens. Actuators A Phys., 1996, 54, 448–456 CrossRef.
- P. Weroński, Y. Jiang and S. Rasmussen, Biophys. J., 2007, 92, 3081–3091 CrossRef.
- P. E. G. Thorén, D. Persson, E. K. Esbjörner, M. Goksör, P. Lincoln and B. Nordén, Biochemistry, 2004, 43, 3471–3489 CrossRef.
- H.-J. Hinz and J. M. Sturtevant, J. Biol. Chem., 1972, 247, 3697–3700 CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Representative Δf-t plots showing overtone effects for PNA1, PNA2, PNA3 and PNA4 (Fig. S1–S4); Δf-t plot for PNA1 on DMPC/DMPG (4:1) lipid membrane (Fig. S5); Energy dissipation (ΔD) vs.frequency (Δf) dependence plots for PNA1, PNA3 and PNA4 on DMPC/DMPG (4:1) membrane (Fig. S6–S8) and PNA1, PNA2, PNA3 and PNA4 on neat DMPC membrane (Fig. S9–S12). See DOI: 10.1039/c2ra20462b/ |
| This journal is © The Royal Society of Chemistry 2012 |