Ultrasonication-assisted direct functionalization of graphene with macromolecules†
Bin
Shen
,
Wentao
Zhai
*,
Dingding
Lu
,
Jing
Wang
and
Wenge
Zheng
*
Ningbo Key Lab of Polymer Materials, Ningbo Institute of Material Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang province, 315201, China. E-mail: wtzhai@nimte.ac.cn; wgzheng@nimte.ac.cn; Fax: +86 0574 8668 5186; Tel: +86 0574 8668 5256
First published on 13th April 2012
Abstract
Poly(vinyl alcohol) (PVA) functionalized graphene (f-G) was prepared by ultrasonication of pristine graphene (p-G) in a PVA aqueous solution. PVA macroradicals formed by sonochemical degradation of the PVA solution were successfully trapped by graphene and grafted onto its surface. This was confirmed by transmission electron microscopy, atomic-force microscopy and 1H NMR measurements. The content of PVA on graphene was estimated to be ∼35%. The f-G could be well dispersed in the PVA matrix by a simple solution mixing and casting procedure. Due to the effective load transfer between f-G and PVA matrix, the mechanical properties of the f-G/PVA films were significantly improved. Compared with the p-G/PVA films, a 12.6% increase in tensile strength and a 15.6% improvement of Young's modulus were achieved by addition of only 0.3 wt% f-G. Moreover, our simple ultrasonication technique could enable us to functionalize graphene with other polymers.
1. Introduction
Graphene, characterized by an atomically thin two-dimensional sheet of sp2 atoms, is regarded as “the thinnest material in the universe” with tremendous potential applications.1,2 Due to its high surface area and high aspect ratio, excellent tensile strength, well-defined thermal conductivity and electrical conductivity, graphene is considered useful in various technological areas, such as electronics, sensors, solar cells, memory devices, hydrogen storage, and polymer composites.3–6 When incorporated appropriately, graphene can dramatically enhance the electrical, physical, mechanical, and barrier properties of polymer composites at extremely low loading.7–10 However, the key point to the successful improvement of their performance is the dispersion of graphene in the host polymers. Substantial efforts are now being made to modify graphene surfaces to improve their processability and the performance of the composites.11–15Currently, chemical functionalization of graphene is always based on the graphene from previously prepared graphene oxide, which has multiple oxygen-containing functionalities, such as hydroxyl and carboxyl groups.16 Most of the functionalization reactions reported in the literature are based on the amidation and esterification of graphene-bound carboxyl groups with organic molecules.17–19 With respect to polymer functionalization of graphene, traditional free radical polymerization, atom transfer radical polymerization (ATRP) and reversible addition-fragmentation chain transfer (RAFT) has been used to functionalize polymer chains to the surface of graphene.20–22 However, the above mentioned schemes for functionalization of graphene usually involve multistep organic syntheses. Sometimes, the preparation protocols are rigorous and time-consuming. The most obvious drawback is that the prepared graphene composites are only on a milligram scale. Therefore, it is still a big challenge for scientists to functionalize graphene in a much simpler and more economical manner.
Ultrasound has found important applications in a diverse range of materials and chemical syntheses.23–26 Both the physical and chemical effects of ultrasound arise from acoustic cavitation: the formation, growth, and collapse of bubbles in liquids irradiated with high intensity ultrasound.23–25,27 Localized hot spots with temperature of ∼5000 K and pressures of hundreds of bars are generated during the bubble collapse within liquid, which can induce some chemical reactions that can't take place under normal conditions. Especially in polymer synthesis, a lot of materials have been prepared through ultrasonic induced radical polymerization of monomers.28,29 Taking advantage of this simple technique, Xu et al.30 achieved polystyrene functionalized graphene by ultrasonic irradiation of graphite in styrene. They found that ultrasonic irradiation could mechanochemically exfoliate graphite into graphene sheets combined with functionalization of graphene with polystyrene chains, which were formed from sonochemically initiated radical polymerization of styrene and could make up 18 wt% of the functionalized graphene.
It is also well accepted that ultrasonic irradiation can induce the degradation of polymers, which was attributed to the acoustic cavitation. Polymer chains near the collapsing bubbles were caught in a high-gradient shear field, causing the polymer segments in this shear field to move at a higher velocity than the chains further away from the collapsing cavity. This relative motion of the polymer segments and solvent produced stress on the polymer chain and caused shearing force,31 which led to the breakage of macromolecular C–C bonds, and formed long chain radicals. The researchers have detected the formation of macroradicals during ultrasonic irradiation of poly(methyl methacrylate), polystyrene, and poly(vinyl alcohol) (PVA) using ESR techniques32–34.
In this work, we propose the synthesis of polymer-functionalized graphene by the direct formation of polymer macroradicals from sonochemical degradation of a polymer solution. While ultrasonic irradiation of graphene in a PVA aqueous solution, PVA macroradicals formed by sonochemical degradation of the PVA solution can be trapped by graphene and grafted onto its surface to form PVA-functionalized graphene (f-G).
2. Experimental
Materials
Pristine graphene (p-G) was prepared according to a vacuum-assisted method described in our previous work (see the ESI†). The C/O ratio of the graphene was 8.1, measured with a Shimadzu Axia-Ultra DLD X-ray photoelectron spectroscopy. Poly(vinyl alcohol) (PVA) was purchased from Aldrich (Mw ∼ 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–186000, 99% hydrolyzed). All other reagents were used as received from Shanghai Experiment Reagent Co., Ltd.
000–186000, 99% hydrolyzed). All other reagents were used as received from Shanghai Experiment Reagent Co., Ltd.
Ultrasonication-assisted synthesis of f-G
The main experimental procedure of the preparation of the samples is illustrated in Scheme 1. The PVA (0.5 g) was dissolved in distilled water (100 ml) at 95 °C and the solution was subsequently cooled to room temperature. Then, p-G (50 mg) was gradually added to the solution under vigorous stirring for 30 min. After thorough deoxygenation for 30 min by bubbling with Ar, the mixture was irradiated by an ultrasonic processor (JY-92 IID, Shanghai Xinzhi) with a high-intensity ultrasonic horn (Titanium, 22 kHz, 300 W) for 120 min under Ar flow (Fig. 1) For comparison, pristine graphene was also irradiated in the absence of PVA using the same conditions. After the mixture was subjected to ultrasonic irradiation for 120 min, a homogeneous dispersion was obtained with a small amount of black sediment. The black sediment can be ascribed to impurities such as nonfunctionalized graphene and functionalized graphene with low PVA grafting density.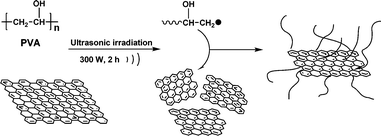 | ||
| Fig. 1 Schematic illustration of the sonochemical preparation process of PVA-functionalized graphene. | ||
The resulting mixture was subjected to low speed centrifugation at 2000 rpm for 20 min. After removal of the black sediment, the black supernatant was first vacuum-filtered through a 0.22 μm Nylon membrane, and the collected black solids were thoroughly washed 3 times with 50 ml hot distilled water. The collected black solids were then redispersed in 100 ml of distilled water by vigorous stirring for 10 min with some heating, filtered, and washed with excess hot distilled water again. This dispersion-filtration-washing cycle was repeated at least 3 times to remove any residual PVA. During each cycle of vacuum-filtration, several drops of filtrate were added into THF solution from time to time, and the absence of cloudiness can be used to indicate the complete removal of PVA. The collected black solids were dried overnight in a vacuum oven at 60 °C. Then, the f-G was obtained. Typically, the yields of the f-G are ∼9% based on the initial graphene used in this sonochemical process.
Preparation of PVA/graphene films
The PVA/graphene thin films were prepared by a solution drop-casting method. First, neat PVA was dispersed in distilled water by stirring at 95 °C for 30 min. Then, p-G and f-G were gradually added into the PVA solution until the content of graphene was about 0.1 wt% and 0.3 wt%. The mixture was stirred for 4–12 h until homogeneous and without any visual air bubbles. The homogeneous solution was then dropped onto a glass substrate for film-casting. The thin film on the substrate was dried at 50 °C for film formation until weight reached an equilibrium value. The thickness of the resulting film was about 60 μm. For tensile tests, the samples were conditioned in the oven (50 °C) for 24 h before testing, as PVA exhibits a high degree of moisture sensitivity. | ||
| Scheme 1 Schematic illustration of the sonochemical preparation process of PVA-functionalized graphene. | ||
Characterization
Raman spectra were excited with a laser of 633 nm and record with Labram spectrometer (Super LabRam II system). All the powder samples were directly deposited on the wafer in the absence of solvent. Transmission electron microscopy (TEM) was conducted on a Tecnai G2 F20 transmission electron microscope with an accelerating voltage of 100 kV. The samples for TEM measurements were prepared by placing one drop of sample dispersion in distilled water on carbon-coated copper grids. Typical tapping-mode atomic-force microscopy (AFM) measurements were performed using Multimode SPM from Digital Instruments with a Nanoscope V Controller from Veeco. The dispersion was deposited onto a silicon plate and dried in the air. 1H NMR measurements were performed on a Bruker Avance III 400 MHz NMR spectrometer. The samples for NMR measurements were prepared by dispersing the sample in DMSO-d6. Thermal gravimetric analyses (TGA) were conducted on Mettler-Toledo TG/DSC 1, with a heating rate of 20 °C min−1 in a nitrogen atmosphere. The crystallinity of the film samples were investigated by differential scanning calorimetry (DSC) using a Mettler-Toledo TG/DSC 1 instrument. The tensile properties of the films were measured on Instron 5567 materials testing system at 23 °C with 35% relative humidity. The extension rate was 5 mm min−1 and the gauge length was ∼50 mm. All samples were cut into strips of ∼150 mm × 10 mm using a razor blade. The thickness of each strip was measured utilizing a low torque digital micrometer. In all cases, more than five samples were tested from which the mean and standard deviation were calculated.3. Results and discussion
As shown in Fig. 1, ultrasonic irradiation of graphene in a PVA aqueous solution produced PVA chain radicals by sonochemical degradation of the PVA solution, which has been reported by other researchers.34 On the other hand, ultrasound could exfoliate few-layer graphene sheets into single-layer graphene sheets and cut them into smaller sheets by the mechanical shockwaves and shear forces created by the collapse of cavitating bubbles.24,35,36 Furthermore, high intensity ultrasound was found to cause serious damage to carbon nanoparticles, such as carbon nanotubes.37 Significant amounts of dislocation defects were observed in the treated nanoparticles and these defects may be vulnerable areas towards subsequent attack by ultrasound cavitation.37–39 Similarly, ultrasonic irradiation of graphene will also produce defects in the surface of graphene, which has been reported by some researchers.35,36 In order to confirm this result, pristine graphene and graphene irradiated by ultrasound for 120 min were studied by Raman spectroscopy, which is shown in Fig. 2. In all samples, two prominent peaks are clearly visible, corresponding to the so-called D and G band at 1335 and 1584 cm−1, respectively.40–42 The D band arises from the activation in the first order scattering process of sp3 carbons, which has been attributed to dislocation defects in graphene sheets.43 And the intensity ratio of D and G bands expresses the sp3/sp2 carbon ratio, a measure of the extent of defects.40 This suggests that the D/G ratio can be used to monitor the extent of defects by ultrasound. For our samples, the D/G ratio of pristine graphene and graphene irradiated by ultrasound for 120 min are respectively 3.0 and 3.4, reflecting the increase in defects after ultrasound. Therefore, ultrasonic irradiation of graphene can cause a considerable amount of defects on the graphene surface, which might produce reactive sites in situ as a result of the high temperature and pressure during bubble collapse. Moreover, the original defects on pristine graphene can also be easily destroyed and produce reactive sites during ultrasonic irradiation. The PVA chain radicals produced by sonochemical degradation of the PVA solution can react easily with graphene, because of the reactive sites formed on the graphene surface, and readily functionalize them via a “grafting to” method. In addition, some researchers have reported that the macroradicals have a high tendency to react with the sp2 hydridized carbon on graphene,30,44,45 thus producing f-G. | ||
| Fig. 2 Raman spectra of (a) pristine graphene and (b) graphene irradiated by ultrasonication for 120 min, with a laser excitation wavelength of 633 nm. | ||
Fig. 3 compares the solubility in water of p-G and f-G. In contrast to p-G, which is insoluble in water (Fig. 3A), f-G prepared after ultrasonic irradiation in PVA solution can be easily dispersed in water (Fig. 3B). Upon addition of the water dispersion of f-G into an excess of THF, which is a poor solvent for PVA, insoluble black solids quickly phase separated out. Thus, by visual inspection, the PVA chains have been successfully grafted onto the surface of graphene. The dispersion of f-G in water is indefinitely stable, and no sedimentation was observed up to the present (more than 2 months). In the control experiment, we mixed p-G and PVA at a weight ratio of 1:1 in water. After storage for ∼1 day, the graphene precipitated out (Fig. 3C). This suggests that the good solubility of f-G is due to the covalent grafting of degraded PVA instead of physisorption of PVA chains onto the surface of graphene.
 | ||
| Fig. 3 Photographs of graphene samples in water: (A) p-G; (B) f-G; (C) the mixture of p-G (50 wt%) and PVA (50 wt%); All photos were taken after 2 months of storage at room temperature. The concentrations of f-G in sample B is 0.2 mg ml−1. | ||
The nanostructures of f-G were investigated by TEM. The TEM image shown in Fig. 4a reveals a transparent clean surface with a few thin ripples for p-G. As reported previously, the ripples are intrinsic properties of thin graphene sheets, which is due to the extra thermodynamic stability of the 2D membranes arising from microscopic crumpling.46 In contrast, the image of f-G is entirely different, which is shown in Fig. 4b. We can clearly observe that the surface of graphene is covered by a thin coating, which are likely to be the grafted PVA polymers. This morphology is similar to the case of polymer-functionalized carbon nanotubes, where a relatively uniform polymer interface layer is created.47,48 AFM was then employed to examine the thickness of f-G. As shown in Fig. 4c, the approximate thickness of p-G sheet is ∼1.2 nm, which is very close to the reported apparent thickness of single graphene.49,50 And the average thickness of the f-G sheet is ∼3.8 nm according to cross-sectional analysis (Fig. 4d), which is much higher than that of p-G. This apparent height difference is probably caused by the grafted PVA on the surface of graphene. Assuming that both sides of the sheets have been grafted, the thickness of the PVA layer grafted on each surface of the graphene sheets can be deduced to be ∼1.3 nm, which is sufficient to functionalize the graphene sheets. In addition to the thickness difference, the presence of grafted PVA chains is further supported by the roughened graphene surface. As a typical representative, the roughness (Rq and Ra) was calculated by the AFM software (Research NanoScope 7.20). The grafted PVA chains on the surface of f-G result in surface roughness (Rq = 0.94 nm, Ra = 0.62 nm) because the functionalization sites via radical coupling on the surface of graphene are randomly distributed on the surface.23 In contrast, p-G always shows a smooth and flat surface (Rq = 0.42 nm, Ra = 0.29 nm).
 | ||
| Fig. 4 TEM images and AFM images of (a) (c) pristine graphene and (b) (d) PVA-functionalized graphene. | ||
The presence of PVA chains on f-G was also examined by 1H NMR, which can provide rich information about the functional groups grafted onto the graphene surface. The 1H NMR spectrum of f-G in DMSO-d6 is compared with that of neat PVA in Fig. 5. The spectrum of f-G exhibits the PVA backbone signals at 3.95–3.75 ppm (CH2C![[H with combining low line]](https://www.rsc.org/images/entities/b_i_char_0048_0332.gif) OH), 1.6–1.2 ppm (C2CHOH) and 5–4 ppm (CH2CHO).47 Upon the attachment to graphene, the f-G maintained similar chemical shifts but became more broadened, which suggests that the PVA chains are associated with diamagnetic species of high molecular weight and low mobility.51 Here, the only possible species in the solution were the graphene sheets. This further supports that the PVA chains had been grafted onto the graphene surface.
OH), 1.6–1.2 ppm (C2CHOH) and 5–4 ppm (CH2CHO).47 Upon the attachment to graphene, the f-G maintained similar chemical shifts but became more broadened, which suggests that the PVA chains are associated with diamagnetic species of high molecular weight and low mobility.51 Here, the only possible species in the solution were the graphene sheets. This further supports that the PVA chains had been grafted onto the graphene surface.
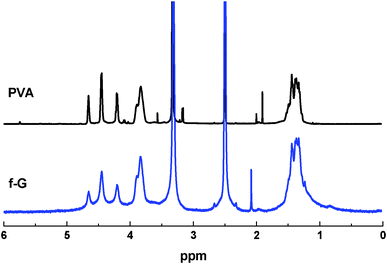 | ||
| Fig. 5 1H NMR spectra of neat PVA and f-G in DMSO-d6. | ||
TGA was carried out to determine the grafted PVA content in the f-G. As shown in Fig. 6, the weight loss of neat PVA reached the equilibrium at a temperature of 600 °C, indicating that all PVA had been burned out. It is noted that the p-G lost 12% of its original weight at 600 °C. By comparing the weight loss of the f-G, p-G and neat PVA, the PVA content in f-G was evaluated to be ∼35%. In order to confirm that the above results were not simply due to the physisorption of the PVA chains onto the graphene surface, the mixture of p-G and PVA with vigorous stirring was prepared as mentioned in Fig. 3C. The dispersion-filtration-washing cycle was repeated at least 3 times to remove any residual PVA on p-G. The TGA result (Fig. S1†) showed that further purification of graphene by the previously described filtration and washing steps led to p-G free of any adsorbed PVA, which indicated that the above results were due to the covalent grafting of degraded PVA instead of physisorption of PVA chains onto the graphene surface.
 | ||
| Fig. 6 TGA curves of p-G, neat PVA and f-G with a heating rate of 20 °C min−1 under N2. | ||
However, it is challenging to directly determine the grafted PVA molecular weight on the graphene surface. As a model experiment, Naake et al.52 reported that the sonochemical-degraded molecular weight of PMMA was independent of the presented radical terminators. Based on this result, we can estimate the molecular weight of degraded PVA by the absence of graphene under the same ultrasonic condition using a viscosity method (see the ESI†). As indicated in Fig. 7, the molecular weight (Mη) of PVA was 64.1 kg mol−1 at the sonication time of 120 min. Furthermore, extending the sonication time from 20 min to 180 min leads Mη to decrease rapidly. Compared with the sonochemical polymerization method reported by Xu et al., our method is much easier to control over the grafted polymer molecular weight on the graphene surface just by adjusting the sonication time. Furthermore, the change of PVA molecular weight might offer us a chance to control the grafted PVA chain length and grated PVA content, which will be studied in the future.
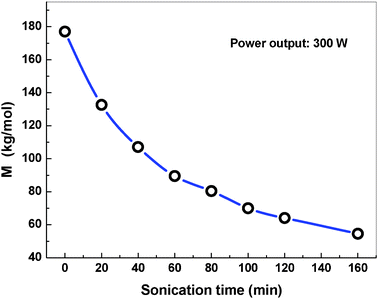 | ||
| Fig. 7 Ultrasonic degradation of PVA in the absence of graphene, Mη as a function of irradiation time. | ||
As an application demonstration, the f-G was used for the preparation of f-G/PVA film by a simple solution mixing and casting procedure. For comparison, the p-G/PVA film was also employed. The films containing 0.3 wt% p-G and f-G were used as examples, and the photographs are shown in Fig. 8b. From the photographs, we can see that the thin films were all of high optical quality and transparent, but the composites film based on f-G exhibited better homogeneity compared to that of p-G. Optical microscope images provide direct evidence for evaluating the dispersion quality of the composite films. Fig. 8a shows images of p-G/PVA and f-G/PVA film, respectively. At micrometer-scale resolution, many dark spots with the diameter of several micrometers were observed in p-G/PVA film, which revealed that the p-G was aggregated seriously in the PVA matrix. By contrast, a homogeneous and uniform f-G/PVA film was observed. The excellent dispersion can be attributed to the improved interface adhesion of f-G with PVA, which can prevent the agglomeration of graphene in PVA matrix.
 | ||
| Fig. 8 Optical micrographs of (a) p-G/PVA and f-G/PVA film and (b) the photograph of PVA/graphene films. | ||
The tensile properties for p-G/PVA and f-G/PVA films were tested after being conditioned in the same environment for 24 h. The typical stress-strain curves are given in Fig. 9 and their mechanical properties are listed in Table 1. Compared with the p-G/PVA film, the tensile strength of the f-G/PVA film, containing 0.3 wt% graphene, increased by 12.6% from 87 to 98 MPa, and Young's modulus increased by 15.6% from 4.5 to 5.2 GPa. The elongation at break of f-G/PVA film was also much higher than that of the p-G/PVA film. Since PVA is a semicystalline polymer, its mechanical properties strongly depend on the degree of crystallinity.53–55 Based on the enthalpy of melting, the crystallinity of the f-G/PVA film was 7.9% lower than that for the p-G/PVA film (Fig. S2†). Therefore, the increased modulus and strength in the f-G/PVA film was not attributed to changes in crystallinity. According to the literature, such mechanical improvements could be attributed to the efficient load transfer between graphene sheets and the polymer matrix.22,56 Here, the PVA chains on the surface of f-G provide stronger interaction with the PVA matrix. As a result, a more effective load transfer across the graphene-PVA interface is obtained, resulting in a significant increase of tensile mechanical properties. Furthermore, the tensile properties of p-G/PVA and f-G/PVA films with 0.1 wt% graphene also presented the same result, which are shown in Table S1†.
 | ||
| Fig. 9 Stress-strain curves for various PVA/graphene films. The content of graphene in the films is about 0.3 wt%. | ||
| Tensile strength (MPa) | Young's modulus (GPa) | Elongation at break (%) | Crystallinity (%) | |
|---|---|---|---|---|
| PVA | 56 ± 3 | 3.4 ± 0.4 | 50 ± 4 | 31.9 |
| p-G/PVA | 87 ± 3 | 4.5 ± 0.2 | 26 ± 6 | 30.2 |
| f-G/PVA | 98 ± 2 | 5.2 ± 0.2 | 38 ± 4 | 22.3 |
In addition to PVA, we tested other polymers, poly(ethylene glycol) (PEG) and poly(vinyl pyrrolidone) (PVP), to explain the applicability of our approach. The dispersion stability experiments were used to estimate whether the polymer chains had been successfully grafted onto the graphene surface. The results are presented in Fig. S3† and Fig. S4†, and showed that ultrasonication of graphene in the PEG and PVP aqueous solution led to the formation of PEG-f-G and PVP-f-G, which was very stable and could be easily dispersed into water. The comparison experiments were also done to confirm that the results were not due to the physisorption of polymer chains onto the graphene surface.
Conclusions
In summary, we developed a facile and efficient technique for functionalization of graphene with PVA. During the process, ultrasonic irradiation was powerful enough to induce the formation of PVA macroradicals and the generation of reactive sites on the graphene surface in PVA aqueous solutions. The PVA macroradicals were grafted onto the graphene surface to form the f-G. Because of the improved interface adhesion of f-G with PVA, the f-G could be well dispersed in the PVA matrix by a simple solution mixing and casting procedure. Due to the effective load transfer between f-G and PVA matrix, the mechanical properties of the f-G/PVA films were significantly improved. Compared with the p-G/PVA films, a 12.6% increase in tensile strength and a 15.6% improvement of Young's modulus were achieved by addition of only 0.3 wt% f-G. Furthermore, the developed ultrasonication functionalization technique has great potential to be extended to other polymer systems.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (Grants 51003115), and Ningbo Natural Science Foundation (Grant No. 2011A6101118) for their financial support of this study.References
- A. K. Geim and A. H. MacDonald, Phys. Today, 2007, 60, 35–41 CrossRef CAS
.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704–1708 CrossRef
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CAS
- R. Miranda and A. L. Vazquez de Parga, Nat. Nanotechnol., 2009, 4, 549–550 CrossRef CAS
- G. Wang, X. Shen, B. Wang, J. Yao and J. Park, Carbon, 2009, 47, 1359–1364 CrossRef CAS
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS
- L. J. Cote, R. Cruz-Silva and J. Huang, J. Am. Chem. Soc., 2009, 131, 11027–11032 CrossRef CAS
- L.-H. Liu, M. M. Lerner and M. Yan, Nano Lett., 2010, 10, 3754–3756 CrossRef CAS
- Y. Zhang, L. Ren, S. Wang, A. Marathe, J. Chaudhuri and G. Li, J. Mater. Chem., 2011, 21, 5386–5391 RSC
- H. J. Salavagione, M. n. A. Gómez and G. Martiínez, Macromolecules, 2009, 42, 6331–6334 CrossRef CAS
- S. Vadukumpully, J. Gupta, Y. Zhang, G. Q. Xu and S. Valiyaveettil, Nanoscale, 2011, 3, 303–308 RSC
- L.-H. Liu and M. Yan, J. Mater. Chem., 2011, 21, 3273–3276 RSC
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720–7721 CrossRef CAS
- M. Quintana, A. Montellano, A. E. del Rio Castillo, G. V. Tendeloo, C. Bittencourt and M. Prato, Chem. Commun., 2011, 47, 9330–9332 RSC
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2010, 20, 1982–1992 RSC
- P. Zhang, K. Jiang, C. Ye and Y. Zhao, Chem. Commun., 2011, 47, 9504–9506 RSC
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS
- K. S. Suslick and G. J. Price, Annu. Rev. Mater. Sci., 1999, 29, 295–326 CrossRef CAS
- W. Zhai, J. Yu and J. He, Polymer, 2008, 49, 2430–2434 CrossRef CAS
- G. Cravotto and P. Cintas, Chem.–Eur. J., 2010, 16, 5246–5259 CAS
- K. S. Suslick and D. J. Flannigan, Annu. Rev. Phys. Chem., 2008, 59, 659–683 CrossRef CAS
- P. Kruus, D. McDonald and T. J. Patraboy, J. Phys. Chem., 1987, 91, 3041–3047 CrossRef CAS
- G. J. Price, D. J. Norris and P. J. West, Macromolecules, 1992, 25, 6447–6454 CrossRef CAS
- H. Xu and K. S. Suslick, J. Am. Chem. Soc., 2011, 133, 9148–9151 CrossRef CAS
- G. Madras and B. J. McCoy, AIChE J., 2001, 47, 2341–2348 CrossRef CAS
- M. Tabata, T. Miyazawa, O. Kobayashi and J. Sohma, Chem. Phys. Lett., 1980, 73, 178–180 CrossRef CAS
- M. Tabata and J. Sohma, Eur. Polym. J., 1980, 16, 589–595 CrossRef CAS
- A. Grönroos, P. Pirkonen, J. Heikkinen, J. Ihalainen, H. Mursunen and H. Sekki, Ultrason. Sonochem., 2001, 8, 259–264 CrossRef
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS
- U. Khan, H. Porwal, A. O'Neill, K. Nawaz, P. May and J. N. Coleman, Langmuir, 2011, 27, 9077–9082 CAS
- K. L. Lu, R. M. Lago, Y. K. Chen, M. L. H. Green, P. J. F. Harris and S. C. Tsang, Carbon, 1996, 34, 814–816 CrossRef CAS
- A. Koshio, M. Yudasaka, M. Zhang and S. Iijima, Nano Lett., 2001, 1, 361–363 CrossRef CAS
- P. J. F. Harris, M. L. H. Green and S. C. Tsang, J. Chem. Soc., Faraday Trans., 1993, 89 Search PubMed
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2007, 8, 36–41 CrossRef
- I. Calizo, A. A. Balandin, W. Bao, F. Miao and C. N. Lau, Nano Lett., 2007, 7, 2645–2649 CrossRef CAS
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238–242 CrossRef CAS
- M. V. Ellacott, L. S. K. Pang, L. Prochazka, M. A. Wilson, J. D. Fitzgerald and G. H. Taylor, Carbon, 1994, 32, 542–544 CrossRef CAS
- M. S. P. Shaffer and K. Koziol, Chem. Commun., 2002, 2074–2075 RSC
- S. Chen, G. Wu, Y. Liu and D. Long, Macromolecules, 2005, 39, 330–334 CrossRef
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS
- Y. Lin, B. Zhou, K. A. Shiral Fernando, P. Liu, L. F. Allard and Y.-P. Sun, Macromolecules, 2003, 36, 7199–7204 CrossRef CAS
- L. Xie, F. Xu, F. Qiu, H. Lu and Y. Yang, Macromolecules, 2007, 40, 3296–3305 CrossRef CAS
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS
- Y.-P. Sun, W. Huang, Y. Lin, K. Fu, A. Kitaygorodskiy, L. A. Riddle, Y. J. Yu and D. L. Carroll, Chem. Mater., 2001, 13, 2864–2869 CrossRef CAS
- G. Schmidt-Naake, M. Drache and M. Weber, Macromol. Chem. Phys., 2002, 203, 2232–2238 CrossRef CAS
- L. Liu, A. H. Barber, S. Nuriel and H. D. Wagner, Adv. Funct. Mater., 2005, 15, 975–980 CrossRef CAS
- X. Zhang, T. Liu, T. V. Sreekumar, S. Kumar, V. C. Moore, R. H. Hauge and R. E. Smalley, Nano Lett., 2003, 3, 1285–1288 CrossRef CAS
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS
- G. Goncalves, P. A. A. P. Marques, A. Barros-Timmons, I. Bdkin, M. K. Singh, N. Emami and J. Gracio, J. Mater. Chem., 2010, 20, 9927–9934 RSC
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2ra01098d |
| This journal is © The Royal Society of Chemistry 2012 |