Chiral optofluidics: gigantic circularly polarized light enhancement of all-trans-poly(9,9-di-n-octylfluorene-2,7-vinylene) during mirror-symmetry-breaking aggregation by optically tuning fluidic media†
Michiya
Fujiki
*a,
Abd Jalil
Jalilah
b,
Nozomu
Suzuki
a,
Makoto
Taguchi
a,
Wei
Zhang
c,
Mohamed Mehawed
Abdellatif
d and
Kotohiro
Nomura
d
aGraduate School of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0101, Japan. E-mail: fujikim@ms.naist.jp; Fax: +81-743-72-6049; Tel: +81-743-72-6040
bSchool of Materials Engineering, Universiti Malaysia Perlis, Kompleks Pusat Pengajian Jejawi 2, 02600 Arau, Perlis, Malaysia. E-mail: jalilahjalil@unimap.edu.my; Fax: +6-04-9798178; Tel: +6-04-9798154
cJiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Chemical Engineering and Materials Science, Soochow University, Suzhou Industrial Park, Suzhou, 215123, China. E-mail: weizhang@suda.edu.cn; Fax: +86-512-658827877; Tel: +86-512-65884243
dDepartment of Chemistry, Graduate School of Science and Engineering, Tokyo Metropolitan University, 1-1 Minami Osawa, Hachioji, Tokyo 192-0397, Japan. E-mail: mehawed_666@yahoo.com; ktnomura@tmu.ac.jp; Fax: +81-42-677-2547; Tel: +81-42-677-2547
First published on 27th June 2012
Abstract
Here we report the first chiral optofluidic system that enables the emergence and enhancement of chiroptical signals of μm-sized polymer particles from achiral all-trans-poly(9,9-di-n-octylfluorene-2,7-vinylene) (PFV) during mirror-symmetry-breaking aggregation due to the optically active fluidic media consisting of (R)-limonene (1R) and (S)-limonene (1S), chloroform and methanol. These results were proven by refractive index (RI) and specific rotation (SR) of the media as well as circular dichroism (CD), optical rotatory dispersion (ORD), circular polarised luminescence (CPL), UV-vis and photoluminescence spectral characteristics of PFV. For comparison, a triple bond linker poly[(9,9-di-n-octylfluoren-2,7-diyl)-alt-yleneethynylene] (PFE) aggregate, which is an analogue of PFV, did not show any CD-signals in the π–π* transition. Gaussian 03 (TD-DFT, B3LYP, 3-21G basis set) calculations of PFV and PFE trimer models suggested that PFV is CD-/CPL-silent helix due to an equal proportion of P- and M-helices in a double-well with a small barrier height, conversely, that PFE is inherently optically inactive due to non-helix conformation in a single-well. Under optimised chiral optofluid with specific RI values of 1.38–1.39, the μm-sized PFV particles showed a great enhancement in Kuhn's dissymmetry amplitudes by ∼1500 times compared to the computed value in helical models of PFV; the gCD at 473 nm reached +0.078 (1S) and −0.104 (1R), and the gCPL at 474 nm attained +0.056 (1S) and −0.077 (1R), while maintaining a high quantum yield (ΦPL) of 75–88% with a short PL lifetime of ∼0.5 ns. For comparison, as for six π-conjugated molecular aggregates with/without H–H repulsions, including trans-stilbene and diphenylacetyelene, with the help of the 1R-/1S-based media, any detectable CD signals were not observed. These results led to the idea that optically active π-conjugated polymers carrying longer alkyl side groups in a double-well potential may be needed to efficiently generate the corresponding optically active aggregates in the chiral tersolvents.
Introduction
Understanding and controlling mirror-symmetry-breaking (MSB) phenomena in living and non-living worlds has been a long-standing issue since the mid-19th century.1 In 1898, Kipping and Pope demonstrated the first deterministic (controlled) mechanism-driven MSB crystallisation from achiral NaClO3.2 Saturated NaClO3 with D-sugar in water preferentially crystallised as the L-form in 40% ee by D-(+)-glucose and 20% ee by D-(+)-mannitol. A century later, Kondepudi et al. found the chance (stochastic) mechanism-driven MSB crystallisation of NaClO3 under stirring without chiral seeding on a macroscopic level.3 In recent years, three deterministic MSB mechanisms in polymer and supramolecular systems under controlled intra- and inter-molecular chirality seeding were established.4–6 However, when the MSB ability in a fluidic media is insufficient due to the weakness of intra- and intra-molecular interactions, a solvent quantity of chiral substances might be required.The first solvent chirality transfer experiment was reported for benzyl and benzophenone dissolved in (2S,3S)-butanediol.7 The second example was poly(n-hexylisocyanate) dissolved in non-racemic chlorinated solvents and its aggregates suspension in chiral and achiral cosolvents.8 These pioneering works prompted us to employ several controlled MSB experiments from optically inactive polysilanes due to the solvent chirality9–11 and optically active polysilane-modified surface.12 Recently, further studies demonstrated that enantiopairs of naturally occurring terpenes and their analogues are candidates for generating optically active polymers and supramolecules in the forms of aggregates and films.13–17
On the other hand, optofluidics has recently been recognized as the conceptually new fusion of integrated optics and microfluidics.18,19 Modern fluidics offers unique μm-scale analogues of solid-state devices with great flexibility because it is easy to tune several optical properties of a fluid and to obtain optically smooth interfaces between the media with immiscible droplets/aggregates. These advantages lead to low-reflection-loss at the smooth interface with the specific Brewster angle θB, which are composed of the aggregates with a high refractive index (RI) and the surrounding optofluidic media with a low RI. Recently, we demonstrated gigantic enhancement in circular dichroism (CD) and circularly polarised luminescence (CPL) amplitudes of ∼5-μm size poly[n-decyl-(S)-2-methylbutylsilane] particles by exactly tuning a single scalar RI value of the surrounding achiral fluid by ∼3000 times compared to the isolated polymer chains in isotropic media.20
Linearly polarized light is characterised by superposition of pseudoscalar RIs: one for left (l-) and the other for right- (r-) circularly polarized light which conveys the angular momentum of integer ± ℏ.21 Ghosh et al. and Silverman et al. independently reported that circularly polarized light could be enhanced by several orders of magnitude compared to isotropic chiral medium using coupled geometry of plural prismatic cuvettes filled with limonene, carvone, and camphorquinone-containing methanol.22 These surprising results stimulated us to apply a chiral optofluidics system that is composed of μm-sized aggregates with a smooth surface made of photoluminescent π-conjugated polymers suspension in the surrounding optically active tersolvent consisting of limonene (Chart 1), chloroform and methanol.
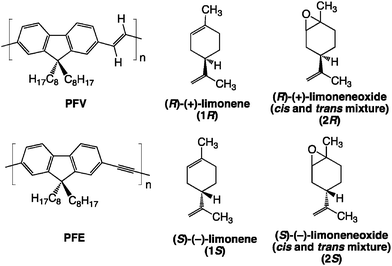 | ||
| Chart 1 Chemical structures of PFV, PFE, limonenes (1R and 1S) and limonene oxides (2R and 2S) used in this work. | ||
Here we demonstrate the greatly enhanced amplitudes in CD, optical rotatory dispersion (ORD) and CPL signals maintaining a high quantum yield (ΦPL) in all-trans-poly(9,9-di-n-octylfluorene-2,7-vinylene) (PFV) during the MSB aggregation in the tersolvent consisting of (R)-limonene (1R) and (S)-limonene (1S), chloroform and methanol. These value were evaluated by the relative method. The absolute ΦPL value in chloroform at room temperature using an integrated sphere was 99%.23 The all-trans-vinylene-linkers and vinyl termini structures were fully characterized as these characteristic 1H-NMR signals (ESI, Fig. S1†). The tersolvent acted as the chiral optofluid because the CD amplitude is maximised at the specific refractive index (RI) and/or optical rotation (OR) of the tersolvent. However, any detectable chiral optofluidic effect for aggregates of poly[(9,9-di-n-octylfluoren-2,7-diyl)-alt-yleneethynylene] (PFE) was not found. Moreover, (R)-limonene oxide (2R) and (S)-antipode (2S) did not provide any such chiral optofluidics effect to the PFV aggregates. We also discuss the origin of the marked difference in the chiral optofluids between PFV and PFE based on Gaussian 03 (TD-DFT, B3LYP, 3-21G basis set) calculations of their trimer models.
Results and discussion
1. CD and CPL chiroptical analysis of polymer aggregates
The CD phenomenon is the differential absorption of left- and right-handed circularly polarised light. This phenomenon can be observed in the electronic absorption bands of any optically active chromophores in the ground state. The dimensionless Kuhn's dissymmetry CD magnitude is characterised as gCD = 2(εL − εR)/(εL + εR), where εL and εR stand for the extinction coefficient for left and right circularly polarised light, respectively.24 However, the CPL phenomenon can detect the stationary-state of chiral luminophores under the photoexcitation. The dimensionless Kuhn's dissymmetry CPL signal is given by gCPL = 2(IL − IR)/(IL + IR), where IL and IR denote the quantum yields of the left-handed CPL and the right-handed CPL under the excitation of unpolarised light, respectively.24 The ideal pure left- and right-handed CD and CPL signals reach |gCD| = 2 and |gCPL| = 2, respectively. Note that the ordinary CD-/CPL-techniques allow the stationary-state of optical isomerism, secondary- and higher-order structures, aggregates and solid films in the ground and photoexcited states to be studied.25When optically active chromophores and luminophores are due to an electric-dipole-allowed transition (edat) origin, the maximum magnitudes of |gCD| and |gCPL|, although they show an intense molar absorptivity and a highly efficient PL efficiency (ΦPL), are limited to less than ∼10−3.24 When the chromophore and luminophore have an electric-dipole-forbidden transition (edft) origin, then the maximum |gCD| and |gCPL| may increase up to ∼10−1, although they show weak absorptivity and an inefficient ΦPL value.24 According to textbooks,25 the aggregates of aromatic hydrocarbons generally quench their edat-origin PL properties due to the formation of sandwich-shaped π–π stacks, leading to the suppression of long-range energy migration. Recent studies have proven that, despite being achiral, aromatic compounds carrying the appropriate substituents and hyperbranched polymers dispersed in solvents do not greatly suppress their inherent PL properties.26
2. Potential energy surfaces of PFV and PFE in the ground and photoexcited states
As for the potential energy surface (PES) of TFV, which is a model trimer of PFV (Chart 2), TFV exists as a mixture of racemic and diastereomeric rotamers in the ground state, including anti-like P-/M- and syn-like P-/M-pairs (Fig. 1a and 1b). This aspect reflects their quadruple-well potential with small energy barrier (EB) values in the ground state. The anti-like P-/M-states are global minima at the dihedral angles of 150°–175° and 185°–210° and are stabilised by 0.4 kcal mol−1 per two trans-vinylene moieties, as compared to those of the syn-like P-/M-pairs, at dihedral angles of 15° and 345°. The values of EB between the anti-like P- and M-states and between the syn-like P- and M-states are as small as ∼2.8 kcal mol−1 and ∼3.2 kcal mol−1, respectively, which are five times those at the thermal energy of 25 °C (0.6 kcal mol−1); therefore, tunnelling between the P- and M-states can occur.25b The achiral anti- and syn-conformations located at 180° and 0° are energetically unstable. The existence of EB in the proton-tunnelling of tropolones in the vibrational transitions in the S0 and S1 states27 and ring puckering of cyclic compounds in the ro-vibrational modes on the order of 1–2 GHz (3–6 cm−1) in the S0 state28 were established experimentally and theoretically. Experimental studies to test the validity of the tunnelling concept in artificial helix polymers appear to be rare.29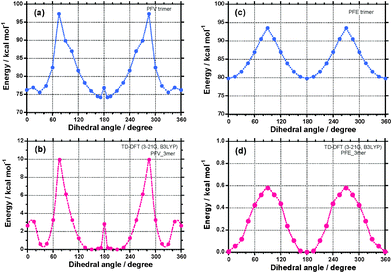 | ||
| Fig. 1 The potential energy surface of TFV ((a) and (b)) and TFE ((c) and (d)) as a function of the dihedral angle. Anti- and syn-conformers (Chart 2) were chosen as 180° and 0°, respectively. (a)/(c) and (c)/(d) are potential energy surfaces in the ground and excited states, respectively. The potential energy surfaces in the excited state (a) and (c) were simply estimated from the difference in the energy between the S0–S1 π–π* transition energy (see, Fig. S2†) and the corresponding potential energy surfaces (b) and (d) in the ground state. | ||
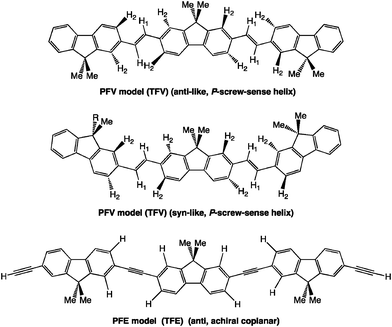 | ||
| Chart 2 PFV and PFE trimer models (TFV and TFE) used for Gaussian 03 calculation (TD-DFT, B3LYP, 3-21G basis set). | ||
The subtle energetic preference (∼0.4 kcal mol−1) of the anti-like TFV for the syn-like TFV is linearly amplified by adding up the number of repeating units, leading to a considerably stabilised anti-like helical PFV. For example, all-anti-like 100-mers would be stabilised by 20 kcal mol−1, as compared to the corresponding all syn-like 100-mers. Note that the twisting ability of TFV originates from the steric demand between the CH protons (H2) of the fluorene rings and the CH protons (H1) of the trans-vinylenes of TFV, while TFE has no such sterically-demanding C–H bonds (Chart 2). The distance between the H1 and H2 in an ideal anti-TFV with a dihedral angle of 180° is 1.88–1.91 Å, which is 0.27–0.30 Å shorter than the van der Waals (vdW) contact of 2 × 1.09 Å = 2.18 Å of two hydrogen atoms.
These results imply that the coexistence of anti-like P- and M-states with dynamically twisting motions in a cooperative manner might lead to non-stationary CD-silent, mirror-symmetry helix—a high entropy state due to conformational freedom—in the ground state. However, the dynamic twisting between the anti-like and syn-like states appears to be impossible because the EB value between the anti- and syn-like states is estimated to be as high as ∼10 kcal mol−1. Actual PFV was regarded as an ideal model of CD-/CPL-silent helical π-conjugated polymer that maintains its free dynamic twisting ability between anti-like P-/M-conformations.
In comparison, TFE, which is a trimer model of PFE, exists stably in the non-helical anti- (dihedral angle 180°) and syn- (dihedral angle 0°) states with an equal probability in both the ground and excited states (Fig. 1c and 1d). There is no difference in the minimum energy between the anti- and syn-states and a small EB value of 0.6 kcal mol−1, suggesting the occurrence of rapid internal twisting between the anti- and syn-conformers. As for PFE, the only difference between TFV and TFE is a linker of fluorene rings, trans-vinylene or acetylene. This similarity implies that, in actual PFE, the coexistence of purely achiral anti- and syn-states is responsible for optical inactivity (and not CD-silent states) in the ground state.
In the photoexcited states, TFV and TFE are expected to adopt PESs similar to the ground states. The PESs were simply estimated from the lowest S0–S1 π–π transition energy (Fig. S2†) by the addition of the corresponding PES in the ground state. TFV has two global minima around the dihedral angles of 165° and 195° and two local minima around the dihedral angles of 15° and 345°. This scenario means that the coexistence of anti-like P- and M-states (and syn-like P- and M-states) should lead to a CPL-silent photoexcited state. The EB value between the anti-like P- and M-states of TFV is ∼2.9 kcal mol−1 per two trans-vinylene sets, supporting the occurrence of efficient tunnelling between the P- and M-states in the excited state. Conversely, TFE exists in both the achiral anti- (dihedral angle 180°) and achiral syn- (dihedral angle 0°) forms with no energetic preference between the excited states. These results infer that the coexistence of the anti- and syn-states of TFE in the excited state should lead to optical inactivity due to the purely achiral excited state.
Actual PFV is assumed to adopt CD-/CPL-silent states in a mirror-symmetry double-well that maintains the freedom of the dynamic twisting ability between anti-like P-/M-conformations in the ground and excited states, while PFE is in optical inactivity due to achiral coplanar conformations in the ground and excited states. Based on these considerations, we hereafter focus on well-established double-well and single-well models rather than the more complex quadruple-well.
The simulated UV-vis (bottom red line) and CD (upper blue line) spectra of two P-TFV with dihedral angles of 172° and 165° by Gaussian 03 (TD-DFT, B3LYP, 3-21G basis set) are given in Fig. S3.† In the case of the 172°-P-TFV, the CD band profile nearly fits with the corresponding UV-vis profile, yielding gCD = −5.2 × 10−5 at 384 nm and gCD = −7.2 × 10−4 at 334 nm; in the case of the 165°-P-PFV around the global minimum, the Cotton CD signals produce a split-type profile, yielding gCD = 9.5 × 10−5 at 382 nm and gCD = −3.7 × 10−3 at 333 nm. These simulations helped us to characterise the enhancement in optical activity of the PFV aggregates induced by the limonene-based optofluid.
By learning from these PESs in the ground and photoexcited states, we can classify optical inactivity into (i) CD-/CPL-silent states due to an equal proportion of P- and M-helices in a double-well and (ii) inherently non-helical state in a single-well. This discrepancy indeed affected the marked difference in the chiral optofluidic characteristics in the following.
3. UV-vis, PL and PLE spectral characteristics of PFV and PFE in chloroform
From the above considerations of PES, the UV-vis, PL and PL excitation (PLE) spectra of PFV (Mw = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, PDI = 1.54) and PFE (Mw = 11700, PDI = 1.57) in chloroform (1.0 × 10−5 mol litre−1 per repeating unit) at 25 °C were measured (Fig. 2). PFV and PFE had a few well-resolved absorption bands as follows: PFV: 0-0′ (456 nm), 0-1′ (429 nm), and 0-2′ (∼401 nm); PFE: 0-0′ (403 nm) and 0-1′ (388 nm). The marked difference, however, was the molar absorptivity (ε) at the 0-0′ band per repeating unit. PFV had intense, sharp 0-0′ bands of ε = 5.74 × 104 and 5.84 × 104, respectively, while PFE had a weaker ε value (∼2.5 × 104) at the 0-0′ band and a relative intensity of the 0-1′ to 0-0′ bands.
000, PDI = 1.54) and PFE (Mw = 11700, PDI = 1.57) in chloroform (1.0 × 10−5 mol litre−1 per repeating unit) at 25 °C were measured (Fig. 2). PFV and PFE had a few well-resolved absorption bands as follows: PFV: 0-0′ (456 nm), 0-1′ (429 nm), and 0-2′ (∼401 nm); PFE: 0-0′ (403 nm) and 0-1′ (388 nm). The marked difference, however, was the molar absorptivity (ε) at the 0-0′ band per repeating unit. PFV had intense, sharp 0-0′ bands of ε = 5.74 × 104 and 5.84 × 104, respectively, while PFE had a weaker ε value (∼2.5 × 104) at the 0-0′ band and a relative intensity of the 0-1′ to 0-0′ bands.
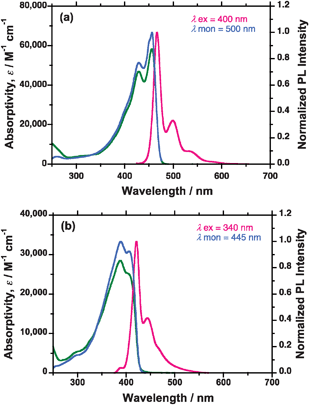 | ||
| Fig. 2 UV-vis, PL and PL excitation (PLE) spectra of (a) PFV (Mw = 27000, and Mw/Mn (PDI) = 1.54) and (b) PFE in chloroform at 25 °C in 1.0 × 10−5 mol L−1 per repeating unit. The green, red and blue lines are the UV-vis, PL and PLE spectra, respectively. | ||
As for the PL spectra, PFV had at least four well-resolved PL bands: 0-0′ (466 nm), 0-1′ (500 nm), 0-2′ (∼537 nm), and 0-3′ (∼581 nm). For comparison, PFE had two resolved PL bands: 0-0′ (421 nm) and 0-1′ (444 nm). These additional satellite bands are due to intense electron–phonon coupling that arises from the discrete quantum energy levels in the ground and excited states.
This situation resulted in a considerably smaller Stokes' shift. When compared to the Stokes' shifts between the 0-0′ bands in the UV-vis and PL spectra, those of PFV and PFE were 10 nm (470 cm−1) and 18 nm (1060 cm−1), respectively. PFV provided a small Stokes' shift, reflected in the main chain regularity and uniformity of the PES curves in the ground and excited states.
4. Enhanced CD-/CPL-amplitudes from μm-sized PFV particles in optofluids
To rationally design chiroptical materials which have great magnitudes of |gCD| and |gCPL| with a high ΦPL, the trade-off relationship should be solved. The most general approach may be by introducing non-racemic chiral side-chains to π-conjugated main chain with a high PL ability.30 To overcome this trade-off, the values of |gCD| and |gCPL| may be enhanced by several orders of magnitude by the mechanism of coupled oscillators between plural chromophores and luminophores.31 Fine tuning of optical parameters with the help of the surrounding media might be further needed.20,22Firstly, typical ambidextrous CD- and CPL-spectra of PFV aggregates in the limonene-based optofluid are shown in Fig. 3. The results were obtained under the optimization, as discussed in the next section. Optical microscopic image (scale bar: 100 μm) of PFV aggregates prepared in tersolvent [chloroform/(1R or 1S)/methanol = 0.3/0.8/1.9 (v/v/v)] at 25 °C showed that they adopted an almost spherical shape with a smooth surface in the fluid and had many particles 5–10 μm in diameter (Fig. 4a). Also, a photograph of turbid PFV in the cuvette under excitation at 365 nm indicated that aggregates exist as suspension in the fluid (Fig. 4b). Evidently, the sign of the Cotton effects was determined only by the limonene chirality, 1S and 1R. The gCD value at 473 nm induced by 1S and 1R had +0.078 and −0.104, respectively. The magnitudes of gCPL at 474 nm induced by 1S and 1R were +0.056 and −0.077, respectively, while the absolute values induced by 1S are somewhat smaller than those induced by 1R. The degree of enhancement in the chiroptical amplitudes increased by ∼1500 times, compared to the computed values in the helical models of PFV (Fig. S3†). It is noted that these gCD and gCPL values are exceptionally high, compared to the CD-/CPL-active π-conjugated polymers,30 with the exception of the values found in thermal annealing processes.
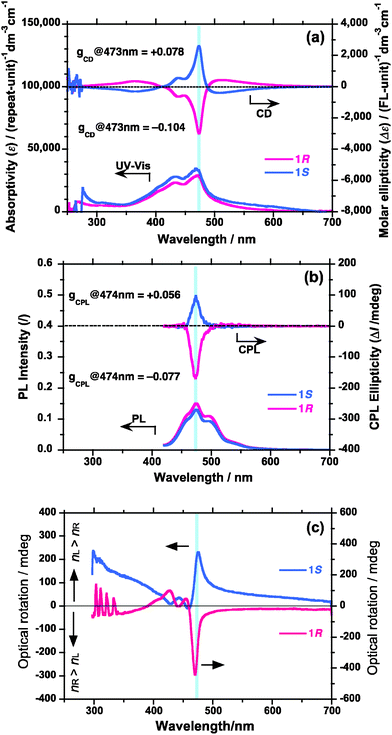 | ||
| Fig. 3 (a) UV-vis/CD, (b) PL/CPL, and (c) ORD spectra of PFV aggregates (Mw = 27000 and Mw/Mn (PDI) = 1.54, 1.0 × 10−5 M) in chloroform/(1R or 1S)/methanol = 0.3/0.8/1.9 (v/v/v) at 25 °C. | ||
 | ||
| Fig. 4 (Left) Optical microscope image (scale bar: 100 μm) of PFV aggregates prepared in tersolvent with chloroform/(1R or 1S)/methanol = 0.3/0.8/1.9 (v/v/v) at 25 °C (scale bar is 100 μm). (Right) Photograph of PFV aggregates dispersed in the tersolvent under excitation at 365 nm. | ||
PFV aggregates in the tersolvent [chloroform/(1S or 1R)/methanol = 0.3/0.8/1.9 (v/v/v)] maintained at a very high ΦPL of 75% (1S) and 88% (1R) (Fig. 5), while the PFV in chloroform had a very high ΦPL of ∼99%. However, the ΦPL value of the PFV aggregates in the cosolvent [chloroform/methanol (0.3/2.7 (v/v)) without limonene dropped to ∼20%. The PL lifetimes of the PFV in chloroform and the PFV aggregates in the tersolvent were 0.43 ns (1S) and 0.46 ns (1R), while the PFV aggregates in the cosolvent had a shorter lifetime of 0.28 ns (Fig. 5 and S4†).
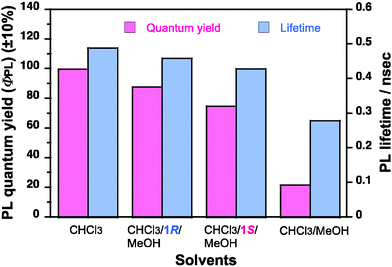 | ||
| Fig. 5 A comparison of the quantum efficiency (ΦPL) and the PL lifetime in ns of PFV in chloroform, aggregates in chloroform/methanol = 0.3/2.7 (v/v), aggregates in chloroform/1R/methanol = 0.3/0.8/1.9 (v/v/v), aggregates in chloroform/1S/methanol = 0.3/0.8/1.9 (v/v/v). | ||
5. Several factors affecting the CD-/CPL-amplitudes
We showed the best results under the optimization in the preceding section. With the aid of clarifying the several factors affecting the emergence and enhancement of chiroptical amplitudes of polymer aggregates by the optically active optofluids, we examined PFV with three different molecular weights and PFE using 1S/1R and 2S/2R, chloroform and methanol based on our two previous outcomes (Chart 1 and S1†).17Of the several polymers in this work, PFV particles with a higher Mw (= 27 000) and narrower Mw/Mn (PDI = 1.54) produced by the limonene-based tersolvent were of particular importance. When the chloroform and total volume of the mixed solvent were fixed at 0.3 mL and 3.0 mL, respectively, then the absolute magnitude of the gCD at the first Cotton band varied greatly with the relative volume fraction of the 1R and methanol (Fig. S5 and S6†). This situation was almost identical to the 1S system that manifested at several tersolvent conditions.
The UV-vis/CD-spectra of PFV in chloroform/methanol = 0.3/2.7 (v/v) and chloroform/(1S or 1R) = 0.3/2.7 (v/v) are shown in Fig. S6a–S6b.† No detectable CD signals were observed in the π–π* transition region of PFV, indicating no P-M preference in the absence of the limonene chirality. As the volume fraction of 1R to methanol changed progressively from 0.3/2.4 to 1.0/1.7, the CD signal from PFV at approximately 470 nm aggregates in the tersolvents and appeared to be due to the 1R chirality origin (Fig. S6c–S6e†). The volume fraction of 1R to methanol was 1.0/1.7, and the CD signal from PFV in the tersolvents at approximately 470 nm abruptly disappeared (Fig. S6f†), though the aggregates were produced according to the naked eye. The greatest gCD values of the PFV particles were obtained using the tersolvent containing chloroform/(1R or 1S)/methanol = 0.3/0.8/1.9 (v/v/v). Hereafter, we chose the tersolvent of chloroform/(1R or 1S)/methanol = 0.3/0.8/1.9 (v/v/v) to test the availability of the limonene tersolvent to PFV and PFE.
The RI values of PFV and PFE in chloroform at four wavelengths (486, 546, 589 and 656 nm) were given in Fig. S7.† The values of optical rotation, α, and RI at 589 nm as functions of volume fractions of 1R/1S, methanol and chloroform were shown in Fig. S8a and 8b.† From these results, the linear relationship between the value of α and RI at 589 nm was obtained in Fig. S8c.† However, the RI value of PFV and PFE in pure chloroform was greatly depended on wavelength.
On the basis of these plots, one can obtain the |gCD| values at the first Cotton band of PFV aggregates as functions of nD and apparent specific rotation ([a]D) in Fig. 6. Evidently, the gCD values maximised at the very specific nD value of 1.38–1.39 and the very specific rotation ([a]D) of the tersolvents, while PFV had RI ∼1.91 at 486 nm. The greatest |gCD| value was ∼8 × 10−2 (equivalent to 4% of the circular polarisation). As for the μm-sized PFV particles under the optimised condition, the enhancement in |gCD| values reached ∼1500 times, compared to the computed value in helical models of PFV. Subtle differences in gCD values between 1R and 1S could be inferable from inherent origin.1
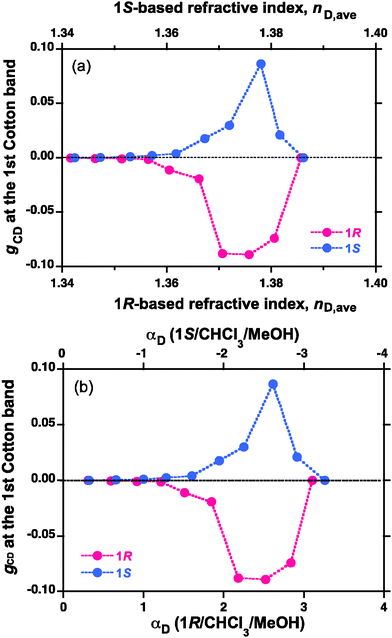 | ||
| Fig. 6 The gCD value of PFV (Mw = 27000, and Mw/Mn (PDI) = 1.54) aggregates as functions of (a) refractive index (nD) at 20 °C and (b) apparent optical rotation (αD per cm at 20 °C) of the chiral tersolvents. | ||
For simplicity, as for unpolarised light, the maximum nrel value of ∼1.384 indicates the Brewster angle θB of ∼54° and refractive angle of 36° (Fig. 7, top). The θB angle indicated no reflection loss for p-polarization and very minimal reflection loss for s-polarization, while a right angle between reflection and refraction light was kept.
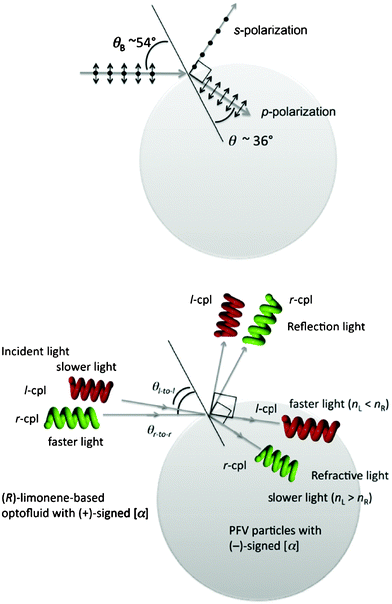 | ||
| Fig. 7 (Top panel) For the unpolarised light model with s- and p-polarizations, incident, reflection and refraction light with θB angle of the chiral particles in the chiral fluid. (Bottom panel) For circularly polarised light model with l- and r-circular polarizations, incident, reflection and refraction light with the hypothetical Brewster angles θB for of the optically active particles in the chiral fluid. Note that, in chiral fluidic media, the wavelength of l-polarised light shortened compared to that of r-polarised light, and, in the chiral particles, wavelength of r-light in the particles shortened compared to that of l-light due to the condition of nL > nR and vice versa, while wavenumbers of these r-/l-light was kept before and after incident light. | ||
However, as for circularly polarised light as incident and reflection modes, fine tunings in the relative nL/nR and/or difference in nL–nR values between the optofluids and optically active PFV aggregates may be crucial to efficiently enhance CD-/CPL-characteristics to confine the angular momentum of +ℏ and −ℏ. The degree of the confinement between +ℏ and −ℏ within the particle is responsible for the great CD-/CPL-enhancement (Fig. 7, bottom).
When compared with optical rotations at 589 nm between the limonene-based tersolvents and optically active PFV aggregates, it is noted that (+)-signed αD in 1R is the opposite of (−)-signed αD in the optically active PFV aggregates induced by the 1R-tersolvent and vice versa. Because the sign of optical rotations means the differences in refractive indices (nL–nR) and speeds of light (vL–vR) between l- and r-circularly polarized light, the l-light slowly travels in the 1R-tersolvent relative to the r-light, conversely, the l-light travels quickly within the aggregates relative to the r-light and vice versa. As illustrated in Fig. 7, bottom, the r-light is then posisble to efficiently confine into the PFV aggregates, leading to enhancement in the negative-sign CD-/CPL-amplitudes. Conversely, when the 1S-based tersolvent was employed, the l-light is confined within the aggregates, providing the positive-sign CD-/CPL-amplitudes.
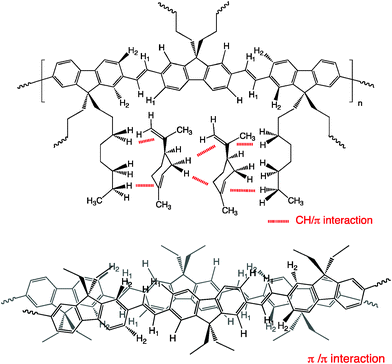 | ||
| Fig. 8 Proposed models of (top) plural intermolecular chiral CH/π interactions between n-octyl chains of PFV and limonene molecules and (bottom) plural intermolecular chiral π/π interactions between PFV in a slipped stacked J-aggregate form. Note that the magnitude of these interactions and detailed stacking structures was small. | ||
Recent vibrational CD (VCD) and DFT calculation results revealed that limonene prefers to adopt three major conformations, with equatorial orientations between the cyclohexene ring and the isopropenyl group.34a The energy difference between three conformers is as small as 0.1 and 0.6 kcal mol−1. Very little energy difference in the n-alkane between the all-trans and gauche-containing conformations suggests that gauche-containing conformations, such as tg, g+g+ and g+g− of n-octyl chains may efficiently wrap limonene molecules by chiral CH/π and vdW interactions.33a
As for π–π interactions, PFV may adopt slip-stacking with a slightly tilted arrangement due to side chain steric interactions, leading to J-aggregates that enable high emission by the edat origin associated with the redshift π–π* transition.35 This phenomenon led to an avoidance of the sandwich-type H-aggregation that cannot emit due to the blueshift of the π–π* transition.35 This idea is evident from the marked redshift of ∼17 nm (790 cm−1) in the 0-0′ π–π* transitions of the PFV aggregates (λmax ∼473 nm) as compared to that in pure chloroform (λmax ∼456 nm) (Fig. 2, 3 and S4†). The redshift of the J-aggregation enables efficient CPL amplitude and PL with a high Φ value due to the edat origin.35
6. Scope and limitation of chiral optofluidics
All-trans-PFV developed by Nomura et al. is a candidate for optical devices because it emits blue light at a nearly 100% ΦPL in solution at room temperature;23 however, the performance might be somehow affected by the polymer structure regularity and uniformity of film, as was already proven for poly(3-alkylthiophene)s in the use of a field-effect transistor.36The optically active PFV aggregates are promising as a solution-processable chiroptical ink because they can efficiently emit blue light at 70–80% ΦPL, which exhibiting the high gCD and gCPL values. However, the limonene protocol is applicable in generating optically active π-conjugated polymer aggregates from the corresponding CD-/CPL-silent helical π-conjugated polymers in a double-well by only tuning the RIs between the optofluids and the polymer particles to satisfy θB. The product yield is near 100% due to the loss-less aggregation method under very mild conditions at ambient temperature in only a few minutes, without any additional thermal treatment for a prolong time.
We tested the possibility of generating optically active aggregates using several molecules without long alkyl groups (Chart S1†) in the limonene-methanol-chloroform tersolvents. In this case, trans-stilbene and oligofluorenes with H–H repulsions are regarded as molecular models of PFV and polyfluorenes, while diphenylacetylene without any H–H repulsion is a molecular model of PFE. Diphenylbithiophene derivative has highly steric repulsions. These molecules have steric repulsions between aryl-H and aryl-H and between aryl-H and vinylene-H excepting for diphenylacetyelene. However, any detectable Cotton CD signals due to π–π* transitions of the aggregates made of these molecules were not found regardless of the presence and absence of the repulsions. This led to an idea that high molecular weight polymers carrying longer alkyl side groups may be needed.
The limonene-based optofluids should provide solution-processable, optically active luminescent aggregates from various achiral polymers.17 Several workers have reported enhanced CD- (and/or CPL-) amplitudes as thin films and aggregates, that are made of π-conjugated polymers with chiral side chains: polythiophene,37 poly(phenylenevinylene),38 poly(p-phenylene),39 poly(p-phenylene-alt-yleneethynylene)40 and polyfluorene family.41 Some of these polymers may enhance their CD-/CPL characteristics by tuning the optofluids.
Experimental section
Details of instrumentation, materials, preparation of optically active polymer aggregates and Gaussian 03 calculation42 are summarised in the ESI.†Conclusion
Here we demonstrated the gigantic CD and CPL amplitudes keeping at a high ΦPL from all-trans-PFV during the MSB aggregation in the tersolvent consisting of 1R and 1S, chloroform and methanol. Because the CD amplitude maximised at the very specific refractive index and optical rotation of the tersolvent, we concluded that the tersolvent acts as the chiral optofluids. However, any detectable chiral optofluidics effect for aggregates of PFE was not found. Gaussian 03 calculations suggested that PFV is a CD-/CPL-silent helix due to an equal proportion of anti-like P- and M-helices in a double-well but PFE is inherently optically inactive due to non-helix conformation in a single-well. The CD-/CPL-silent helix was assumed to be crucial to efficiently emerge chiral optofluidic effects.Acknowledgements
We are grateful for the funds from a Grant-in-Aid for Science Research (22350052), International Cooperation of the Ministry of Science and Technology of China (No. 2011DFA50530) and the National Nature Science Foundation of China (No. 21104052). We thank Kenji Takamizu (Tokyo Metropolitan University: TMU) for GPC/1H-NMR characterisation of polymer samples. We also thank Prof. Hiroshi Masuhara, Prof. Teruki Sugiyama, Prof. Jun-ichi Kikuchi, Prof. Takashi Matsuo, Dr Yoko Nakano, Yoshifumi Kawagoe, Hiroya Fukuda, Ayako Nakao and Kana Yoshida for fruitful discussions, technical guidance and valuable comments. We thank Yasuo Okajima (NAIST) for measuring the PL photodynamics of the PFV and PFE samples.References
- (a) S. F. Mason, Nature, 1984, 311, 19–23 CrossRef CAS; (b) V. A. Avetisov, V. I. Goldanskii and V. V. Kuz'min, Phys. Today, 1991, 44, 33–41 CrossRef CAS; (c) W. A. Bonner, Chirality, 2000, 12, 114–126 CrossRef CAS; (d) B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef; (e) M. Quack, Angew. Chem., Int. Ed. Engl., 1989, 28, 571–586 CrossRef; (f) R. Plasson, D. K. Kondepudi, H. Bersini, A. Commeyras and K. Asakura, Chirality, 2007, 19, 589–600 CrossRef CAS; (g) G. H. Wagniére, On Chirality and the Universal Asymmetry–Reflections in Image and Mirror Image, Wiley-VCH, Weinheim, 2007 Search PubMed; (h) K. Soai and T. Kawasaki, Top. Curr. Chem., 2008, 284, 1–33 CrossRef CAS; (i) J. Crusats, D. Hochberg, A. Moyano and J. M. Ribó, ChemPhysChem, 2009, 10, 2123–2131 CrossRef CAS; (j) A. Guijarro and M. Yus, The Origin of Chirality in The Molecules of Life, RSC, London, 2009 Search PubMed; (k) M. Fujiki, Chem. Rec., 2009, 9, 271–298 CrossRef CAS.
- F. S. Kipping and W. J. Pope, J. Chem. Soc. Trans., 1898, 73, 606–617 RSC.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1995, 267, 832–833 Search PubMed.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CAS.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS.
- B. Bosnich, J. Am. Chem. Soc., 1967, 89, 6143–6148 CrossRef CAS.
- (a) M. M. Green, C. Khatri and N. C. Peterson, J. Am. Chem. Soc., 1993, 115, 4941–4942 CrossRef CAS; (b) C. A. Khatri, Y. Pavlova, M. M. Green and H. Morawetz, J. Am. Chem. Soc., 1997, 119, 6991–6995 CrossRef CAS.
- M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polymer, 2003, 44, 5477–5495 CrossRef.
- H. Nakashima, J. R. Koe, K. Torimitsu and M. Fujiki, J. Am. Chem. Soc., 2001, 123, 4847–4848 CrossRef CAS.
- P. Dellaportas, R. G. Jones and S. J. Holder, Macromol. Rapid Commun., 2002, 23, 99–103 CrossRef CAS.
- A. Saxena, G. Guo, M. Fujiki, Y. Yang, A. Ohira, K. Okoshi and M. Naito, Macromolecules, 2004, 37, 3081–3083 CrossRef CAS.
- (a) S. J. George, Ž. Tomović, M. M. J. Smulders, T. F. A. de Greef, P. E. L. G. Leclère, E. W. Meijer and A. P. H. J. Schenning, Angew. Chem., Int. Ed., 2007, 46, 8206–8211 CrossRef CAS; (b) K. Toyofuku, Md. A. Alam, A. Tsuda, N. Fujita, S. Sakamoto, K. Yamaguchi and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 6476–6480 CrossRef CAS.
- N. Katsonis, H. Xu, R. M. Haak, T. Kudernac, Z. Tomović, S. George, M. Van der Auweraer, A. P. H. J. Schenning, E. W. Meijer, B. L. Feringa and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 4997–5001 CrossRef CAS.
- J. Aimi, K. Oya, A. Tsuda and T. Aida, Angew. Chem., Int. Ed., 2008, 47, 5153–5156 CrossRef CAS.
- (a) A. M. Buono, I. Immediata, P. Rizzo and G. Guerra, J. Am. Chem. Soc., 2007, 129, 10992–10993 CrossRef CAS; (b) P. Rizzo, M. Beltrani and G. Guerra, Chirality, 2010, 22, E67–E73 CrossRef CAS.
- (a) Y. Nakano, Y. Liu and M. Fujiki, Polym. Chem., 2010, 1, 460–469 RSC; (b) Y. Kawagoe, M. Fujiki and Y. Nakano, New J. Chem., 2010, 34, 637–647 RSC; (c) W. Zhang, K. Yoshida, M. Fujiki and X. Zhu, Macromolecules, 2011, 44, 5105–5111 CrossRef CAS; (d) Y. Nakano, F. Ichiyanagi, M. Naito, Y. Yang and M. Fujiki, Chem. Commun., 2012, 48, 6636–6638 RSC.
- (a) D. Psaltis, S. R. Quake and C. Yang, Nature, 2006, 442, 381–386 CrossRef CAS; (b) C. Monat, P. Domachuk and B. J. Eggleton, Nat. Photonics, 2006, 1, 106–114 CrossRef.
- (a) J. A. Sherwin and A. Lakhtakia, Opt. Commun., 2002, 214, 231–245 CrossRef CAS; (b) J. Pedersen and N. A. Mortensen, Appl. Phys. Lett., 2007, 91, 213501 CrossRef; (c) N. A. Mortensen and S. Xiao, Appl. Phys. Lett., 2007, 90, 141108 CrossRef; (d) F. B. Arango, M. B. Christiansen, M. Gersborg-Hansen and A. Kristensen, Appl. Phys. Lett., 2007, 91, 223503 CrossRef; (e) H. C. Hunt and J. S. Wilkinson, Microfluid. Nanofluid., 2008, 4, 53–79 CrossRef CAS.
- Y. Nakano and M. Fujiki, Macromolecules, 2011, 44, 7511–7519 CrossRef CAS.
- (a) R. A. Beth, Phys. Rev., 1936, 50, 115–125 CrossRef; (b) A. H. S. Holbourn, Nature, 1936, 137, 31–31 CrossRef; (c) M. N. Saha and Y. Bhargava, Nature, 1931, 128, 870 CrossRef CAS.
- (a) A. Ghosh and P. Fischer, Phys. Rev. Lett., 2006, 97, 173002 CrossRef; (b) M. P. Silverman, J. Badoz and B. Briat, Opt. Lett., 1992, 17, 886–888 CrossRef CAS.
- (a) K. Nomura, N. Yamamoto, R. Ito, M. Fujiki and Y. Geerts, Macromolecules, 2008, 41, 4245–4249 CrossRef CAS; (b) N. Yamamoto, R. Ito, Y. Geerts and K. Nomura, Macromolecules, 2009, 42, 5104–5111 CrossRef CAS.
- H. P. J. M. Dekkers, in Circular Dichroism: Principles and Applications, ed. N. Berova, K. Nakanishi and R. W. Woody, Wiley–VCH, New York, 2nd edn, 2000, ch. 7 Search PubMed.
- (a) F. Hund, Z. Phys., 1927, 43, 805–826 CrossRef CAS; (b) R. Janoschek, Theories on the Origin of Biomolecular Homochirality, in Chirality–From Weak Bosons to the α-Helix, ed. R. Janoschek, Springer, Berlin, Germany, 1992, ch. 2, pp. 18–33 Search PubMed.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Heidelberg, 3rd edn, 2006 Search PubMed; (b) N. J. Turro, Modern Molecular Photochemistry, University Science Books, Mill Valley, 1991 Search PubMed; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- (a) A. C. P. Alves and J. M. Hollas, Mol. Phys., 1972, 23, 927–945 CrossRef CAS; (b) Y. Tomioka, M. Ito and N. Mikami, J. Phys. Chem., 1983, 87, 4401–4405 CrossRef CAS; (c) F. A. Ensminger, J. Plassard and T. S. Zwier, J. Phys. Chem., 1993, 97, 4344–4353 CrossRef CAS; (d) H. Sekiya, T. Tsuji, S. Ito, A. Mori, H. Takeshita and Y. Nishimura, J. Chem. Phys., 1994, 101, 3464–3471 CrossRef CAS.
- J. Laane, in Frontiers of Moleular Specroscopy, ed. J. Laane, Elsevier, Boston, 2009, ch. 4 Search PubMed.
- M. Fujiki, J. R. Koe, H. Nakashima, M. Motonaga, K. Terao and A. Teramoto, J. Am. Chem. Soc., 2001, 123, 6253–6261 CrossRef CAS.
- (a) G. Bidan, S. Guillerez and V. Sorokin, Adv. Mater., 1996, 8, 157–160 CrossRef CAS; (b) B. M. W. Langeveld-Voss, R. A. J. Janssen, M. P. T. Christiaans, S. C. J. Meskers, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1996, 118, 4908–4909 CrossRef CAS; (c) M. M. Bouman and E. W. Meijer, Adv. Mater., 1997, 7, 385–387 CrossRef; (d) B. M. W. Langeveld-Voss, R. J. M. Waterval, R. A. J. Janssen and E. W. Meijer, Macromolecules, 1999, 32, 227–230 CrossRef CAS; (e) Z. B. Zhang, M. Fujiki, M. Motonaga and C. E. McKenna, J. Am. Chem. Soc., 2003, 125, 7878–7881 CrossRef CAS.
- (a) H. Fröhrich, Phys. Rev., 1950, 79, 845–846 CrossRef; (b) P. Andrew and W. L. Barnes, Science, 2000, 290, 785–788 CrossRef CAS; (c) H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539 CrossRef CAS; (d) J. P. S. Farinha and J. M. G. Martinho, J. Phys. Chem. C, 2008, 112, 10591–10601 CrossRef CAS; (e) I. Hwang and G. D. Scholes, Chem. Mater., 2011, 23, 610–620 CrossRef CAS.
- (a) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University Press: Oxford, 2001 Search PubMed; (b) P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS; (c) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS.
- (a) F. P. Ureña, J. R. A. Moreno and J. J. L. González, Tetrahedron: Asymmetry, 2000, 20, 89–97 Search PubMed; (b) H. Rhee, Y.-G. June, J.-S. Lee, K.-K. Lee, J.-H. Ha, Z. H. Kim, S.-J. Jeon and M. Cho, Nature, 2009, 458, 310–313 CrossRef CAS; (c) S. Abbate, G. Longhi, L. Ricard, C. Bertucci, C. Rosini, P. Salvadori and A. Moscowitz, J. Am. Chem. Soc., 1989, 111, 836–840 CrossRef CAS; (d) E. D. Lipp and L. A. Nafie, Appl. Spectrosc., 1984, 38, 774–778 CrossRef CAS.
- (a) J. B. Klauda, R. W. Pastor and R. B. Brooks, J. Phys. Chem. B, 2005, 109, 15684–15686 CrossRef CAS; (b) V. Pophristic and L. Goodman, Nature, 2001, 411, 565–568 CrossRef CAS; (c) H. Kikuchi, K. Morihashi and O. Kikuchi, J. Chem. Phys., 1998, 108, 2041–2043 CrossRef.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. H. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- (a) G. Bidan, S. Guillerez and V. Sorokin, Adv. Mater., 1996, 8, 157–160 CrossRef CAS; (b) B. M. W. Langeveld-Voss, R. A. J. Janssen, M. P. T. Christiaans, S. C. J. Meskers, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1996, 118, 4908–4909 CrossRef CAS.
- E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909–9910 CrossRef CAS.
- (a) R. Fiesel and U. Scherf, Acta Polym., 1998, 49, 445–449 CrossRef CAS; (b) H. Goto and K. Akagi, Angew. Chem., Int. Ed., 2005, 44, 4322–4328 CrossRef CAS.
- (a) R. Fiesel and U. Scherf, Macromol. Rapid Commun., 1998, 19, 427–431 CrossRef CAS; (b) J. N. Wilson, W. Steffen, T. G. McKenzie, G. Lieser, M. Oda, D. Neher and U. H. F. Bunz, J. Am. Chem. Soc., 2002, 124, 6830–6831 CrossRef CAS.
- (a) M. Oda, H. G. Nothofer, G. Lieser, U. Scherf, S. C. J. Meskers and D. Neher, Adv. Mater., 2000, 12, 362–365 CrossRef CAS; (b) H.-Z. Tang, M. Fujiki and M. Motonaga, Polymer, 2002, 43, 6213–6220 CrossRef CAS; (c) G. Lakhwani and S. C. J. Meskers, Macromolecules, 2009, 42, 4220–4223 CrossRef CAS; (d) R. Abbel, A. P. H. J. Schenning and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4215–4233 CrossRef CAS; (e) J.-M. Yu, T. Sakamoto, K. Watanabe, S. Furumi, N. Tamaoki, Y. Chen and T. Nakano, Chem. Commun., 2011, 47, 3799–3801 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, materials, preparation of polymer aggregates, Gaussian 03 calculation, 1H-NMR spectrum and assignment of PFV, the theoretical S0–S1 and S0–T1 transition energy of TFV and TFE as a function of dihedral angles, simulated UV/CD spectra of helical TFV, PL dynamics of PFV and PFE, the gCD value of PFV as functions of fractions of methanol/limonene and enantiopurity of limonene, UV-vis and CD spectra of PFV and PFE (1.0 × 10−5 M) in the chiral tersolvent and cosolvent at 25 °C, and optical rotation and refractive index of the tersolvents, and refractive index of PFV and PFE. See DOI: 10.1039/c2ra20430d |
| This journal is © The Royal Society of Chemistry 2012 |