Cubic mesophase nanoparticles doped with superparamagnetic iron oxide nanoparticles: a new class of MRI contrast agent
Durga P.
Acharya
*a,
Bradford A.
Moffat
b,
Anastasios
Polyzos
a,
Lynne
Waddington
c,
Greg
Coia
a,
David K.
Wright
d,
Hong X.
Wang
d,
Gary F.
Egan
d,
Benjamin W.
Muir
*a and
Patrick G.
Hartley
a
aCSIRO Materials Science and Engineering, Bayview Avenue, Clayton, VIC 3168, Australia
bThe University of Melbourne, Department of Radiology, Parkville, VIC 3050, Australia
cCSIRO Materials Science and Engineering, 343 Royal Parade, Parkville, VIC 3050, Australia
dHoward Florey Institute, The University of Melbourne, VIC 3010, Australia. E-mail: Durga.Acharya@csiro.au; Tel: +61 3 9545 2518Ben.Muir@csiro.au; Tel: +61 3 9545 2452
First published on 26th June 2012
Abstract
The ability of superparamagnetic iron oxide nanoparticles (SPIONs) to shorten the effective transverse relaxation time (T2) during magnetic resonance imaging (MRI) makes them excellent contrast agents in diagnostic applications. Here we describe a new class of hybrid MRI contrast agent using dispersions of lyotropic bicontinuous cubic phase nanoparticles doped with SPIONs. Hybrid mesophase nanoparticles (HMNs) combining the cubic order of a lyotropic lipid system and SPIONs were successfully prepared and characterized. Highly monodisperse 8 nm spherical SPIONs coated with oleic acid were dispersed in the bulk cubic phase forming lipid matrix of phytantriol nanoparticles 180 nm in size. Transverse relaxivity (r2) measurements show that enhancement of the T2 relaxation time of the HMNs is proportional to the loading of SPIONs in the mesophase nanoparticles. Excellent contrast enhancement in T2 weighted images in the kidney and liver of live rats was observed after intravenous injection of the hybrid mesophase nanoparticles. Results indicate that the HMNs are rapidly transported to the renal system making them useful for contrast enhancement of renal and hepatic systems.
Introduction
Superparamagnetic iron oxides nanoparticles (SPIONs) are routinely used in the clinic as magnetic resonance imaging (MRI) contrast agents and are the subject of a large amount of ongoing research to optimise their material properties for improved performance in vivo.1–4 The unique superparamagnetic properties of SPIONs arise from a combination of factors including atomic composition, surface chemistry, crystal/grain structure, size/shape and mode of dispersion (monodisperse, aggregated etc). Owing to a high magnetization, which is approximately one order of magnitude greater than those of paramagnetic particles, these SPIONs can locally offset external static magnetic fields and disturb their homogeneity on a microscopic scale.SPIONs work by influencing both the longitudinal or spin–lattice relaxation time (T1) and transverse or spin–spin relaxation time (T2) of water protons located in close proximity to the particles on application of an external magnetic field. However, due to their predominant effect of shortening the T2 relaxation time, a negative contrast enhancement in T2 weighted images is observed. The consequence of this effect is to significantly darken areas with high concentrations of SPIONs in T2 and T2* weighted magnetic resonance (MR) images where the time constant T2* includes the contribution of the inhomogeneity in static magnetic fields and the spin–spin relaxation on the body. These particles can also be used as positive contrast agents by choosing appropriate imaging sequence algorithms.5,6 The presence of a large number of iron atoms within each SPION and their high magnetic moment provide strong signal intensity enhancement per unit of metal. Their in vivo performance can be improved via new synthesis techniques that enhance their magnetic properties through control of the iron crystal phases formed.7
The desirable features of SPIONS for use as MRI contrast agents include their ‘in general’ biodegradability and ‘in general’ biocompatibility, ease of synthesis/characterization, and the ability to modify their magnetic properties.8 However there remain clinical challenges relating to suboptimal biodistribution and a short blood (plasma) half life9 which limits the ‘imaging window’.
SPIONs can be produced using organic and aqueous solvent techniques. Although the use of organic solvents generally allows for greater control over nanoparticle size, shape and crystal structure,10 this method results in hydrophobic SPIONs that are capped with organic lipid ligands. The resulting hydrophobic particles do not readily undergo phase transfer, allowing dispersion in water, which is required for biocompatibility. Hence clinically approved SPIONs are synthesized in water and dispersed through the addition of amphiphilic polymers such as dextran resulting in the formation of agglomerates of multiple SPIONs.11 Recent research efforts have been aimed at developing new phase transfer chemistries to solubilise these particles for use in vivo.12,13 Traditionally, the approach used is to sterically or charge stabilize SPIONs with polymers or small molecules that either bind through metal coordination of the iron oxide cores14 or through hydrophobic interactions with lipid groups on the SPIONs.
Another approach to address the challenges of phase transfer and dispersal of SPIONs has been the use of lipid–based colloidal aggregates as the dispersants or carriers. The concept of using liposomes or lamellar phases doped with magnetic nanorods has been previously reported.15–18 In this case, the nanoparticles reside within the lipid bilayers of the vesicles. However, the thin membranes of liposomes made them poorly stable and thus may limit their use as delivery vehicles in vivo.19 We proposed that use of alternative lipid bilayer structures such as colloidal dispersions of lipid mesophases may overcome the limitations associated with liposomes20 due to their complex nanostructure which may allow for increased loading of SPIONs.
Bicontinuous cubic mesophases formed by some polar lipids such as glycerol monooleate and phytantriol consist of interwoven polar and nonpolar channels and therefore can solubilize both hydrophilic and hydrophobic molecules. The mesophase can be dispersed into submicron particles (also called cubosomes™)21–24 in water by mechanical processing. These particles exhibit thermodynamic stability in buffer and have large surface areas (400 m2 g−1) thereby potentially permitting a high loading density for the introduction of SPIONs. In addition, these mesophase nanoparticles exhibit low cytotoxicity in cell based assays and suitable pharmacokinetic behaviour in animal tests which make them potentially useful in biologically relevant applications such as delivery of drugs, bioactives, neutraceuticals and diagnostic agents.25–32 Due to their small particle size (∼ 200 nm), low viscosity, biocompatibility and thermodynamic stability in excess water, these nanoparticles are particularly suitable for intravenous injection. They allow for the incorporation of multiple components such as targeting ligands, fluorescent molecules and therapeutic agents, offering the potential to act as tissue targeting and multimodal imaging and therapeutic agents.33–37 Recently, Boyd et al. reported the phase transfer of hydrophobic gold nanorods into aqueous dispersion of lipidic mesophase submicron particles.38
A number of reports have investigated methods of surface functionalising hydrophobic SPIONs with hydrophilic ligands such as polymers and small molecules to make them colloidally stable in water39–41 and occasionally buffer. In this work, we explore the use of a different approach by using bicontinuous cubic mesophase forming nanoparticles. This approach offers several advantages such as the ability to incorporate multimodal imaging agents and targeting ligands, drug and pro-drugs, dyes, hydrophilic and hydrophobic materials. The method to produce such biocompatible lipidic materials is simple and scalable which makes them potentially useful for a number of therapeutic applications. In this work we report the preparation of hybrid mesophase nanoparticles (HMNs) loaded with SPIONs, their characterization, high throughput relaxivity measurements and MRI contrast measurements in rats to assess their performance as a contrast agent.
Experimental
Materials
Phytantriol (3,7,11,15-Tetramethylhexadecane-1,2,3-triol, BASF) was used as received without further purification. PEO-PPO-PEO type triblock copolymer Pluronic F-127 (Sigma) was used as received. Chloroform (spectroscopic grade, Sigma) and Millipore Milli Q water were also used in this work.Preparation of SPIONs
SPIONs were produced by a modified method published first by Park et al.10 Sodium hydroxide (4.8 g, 120 mmol) was dissolved in 50 mL of distilled water and 80 mL of ethanol was then added. Oleic acid (40 mL, 120 mmol) was slowly added to the mixture solution with stirring and adjusted to pH 7.0. Iron chloride (FeCl3·6H2O, 10.8 g, 40 mmol) was dissolved in 10 mL of distilled water and added to the above mixture, and then 140 mL of hexane was added. The resulting solution was heated to 70 °C for 4 h. When the reaction was completed, the upper organic layer containing the iron-oleate complex was washed three times with 30 mL of distilled water in a separation funnel. After washing, hexane was removed by evaporation, resulting in the formation of a waxy solid. To produce monodisperse iron oxide nanoparticles with a particle size of 8 nm used for this work, 7.2 g (8 mmol) of the iron-oleate complex and 1.14 g of oleic acid (4 mmol, Aldrich, 90%) were dissolved in 40 g of 1-octadecene (Aldrich, 90%) at room temperature. The reaction mixture was first heated to 100 °C and degassed for 30 min, then heated to 320 °C in 50 min and kept at that temperature for 15 min. The resulting brown-black solution containing the nanoparticles was then cooled to room temperature. The iron oxide nanoparticles were precipitated out of 1-octadecene by acetone and then redispersed in chloroform. This washing process was repeated six times to obtain pure SPIONs as a solid powder.In order to produce water soluble SPIONs as a comparison to those inside HMNs the phase transfer of the oleate-capped nanoparticles were carried out by coating them with a commercial polymer using procedure described elsewhere.46 In brief, a solution containing 10 mg of SPIONs in 5 mL of CHCl3 was added to 3.5 mL of a stock solution (1 mg mL−1) of poly(maleic anhydride-co-octadecene) in CHCl3 and the solution was left to stir for 72 h. Then 20 mL of an aqueous 2 M ethanolamine was added to it and left to stir for 24 h until the organic phase became clear. The aqueous phase which contained SPIONs was removed, filtered through a 0.2 μm syringe filter, purified by dialysis against deionized water for 24 h and finally subjected to repeated centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to remove excess polymer from the SPION solution.
000 rpm to remove excess polymer from the SPION solution.
Preparation of cubosomes
One gram of phytantriol and the required amount of SPION (20 mg per gram of phytantriol) were slowly dissolved in 4 mL of chloroform. The solvent was then completely removed by evaporating it in a Buchi Rotavapour at low pressure and elevated temperature (∼ 60 °C). The solvent-free mixture of phytantriol and SPIONs was transferred in to a test tube and a pluronic polymer stabiliser, F-127, was added at a weight fraction of F-127 of 0.15 in the mixture. The mixture was heated at 70 °C for 5 min, followed by vortex mixing and this process was repeated 5 times to ensure that all F-127 was dissolved and homogeneously mixed. This hot mixture was added dropwise to water (at ∼ 65 °C) while applying shear generated by a shear homogenizer (Polytron, Kinematica AG) with a 20 mm probe rotating at 20000 rpm. The dispersion was further homogenized at 65 °C with 3 passes in a pressure homogenizer (EmulsiFlex C5, Avastin) to obtain the final product. The fraction of non aqueous components in the solution of SPIO-loaded lipid nanoparticles was determined by thermogravimetric analysis (Mettler). Approximately 35 mg of nanoparticle solution was taken in 70 μL silica crucible and analysed in the range of 25 to 800 °C. The average of three results was taken to determine the fraction of nonaqueous components in the solution and the concentration of phytantriol in the cubosome solution.
Determination of iron concentration
Iron concentration was obtained by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a Varian Vista ICP-OES spectrometer. Samples for ICP-OES were prepared by heating 300 μL of solution with 700 μL of conc. HNO3 at 75 °C for 3 h. The digested mixture was left at room temperature overnight, then diluted to 20 mL with water and filtered through a 0.45 μm syringe filter before analysis. The average of three separate solutions per nanoparticle batch was taken to determine total iron content of each nanoparticle formulation. The amount of SPIONs loading in cubic phase nanoparticle is expressed in wt% of Fe in phytantriol.Particle size distribution analysis
Particle size distribution measurements were performed in a Zetasizer-Nano instrument (Malvern, UK). Particle size was measured in Milli-Q water using samples appropriately diluted to 1 mg ml−1. The analysis was performed at 25 °C and for each sample, the mean diameter of three determinations was calculated. The data was analysed using the CONTIN algorithm.42–44Cryogenic transmission electron microscopy (Cryo-TEM)
A laboratory-built humidity-controlled vitrification system was used to prepare the nanoparticles for imaging in a thin layer of vitrified ice using cryo-TEM. Humidity was kept close to 80% for all experiments, and ambient temperature was 22 °C. Copper grids (200 mesh) coated with perforated carbon film (Lacey carbon film: ProSciTech, Kirwan, Australia) were used for all experiments. Aliquots (4 μL) of the sample were pipetted onto each grid prior to plunging. After 30 s adsorption time the grid was blotted manually using Whatman 541 filter paper for ∼2 s. Blotting time was optimized for each sample. The grid was then plunged into liquid ethane cooled by liquid nitrogen. Frozen grids were stored in liquid nitrogen until required. The samples were examined using a Gatan 626 cryoholder (Gatan, Pleasanton, CA) and Tecnai 12 transmission electron microscope (FEI, Eindhoven, The Netherlands) at an operating voltage of 120 kV. At all times low-dose procedures were followed, using an electron dose of 8–10 e−/Å2 for all imaging. Images were recorded using a Megaview III CCD camera and AnalySIS camera control software (Olympus.) using magnifications in the range 60000–110000×.
Small-angle X-ray scattering (SAXS)
Scattering experiments were performed on the SAXS/WAXS beamline at the Australian Synchrotron. Glass capillaries (1.5 mm) containing sample solutions were placed in a temperature controlled sample holder maintained at 25 °C by a recirculating water bath. Samples were exposed to the 12 keV X-ray beam and diffraction patterns were recorded using a Pilatus 1 M detector (Dectris, Switzerland). A silver behenate standard was used to calibrate the reciprocal space vector for analysis. Data reduction (calibration and integration) was performed using SAXS15ID program developed at ChemMat CARS, The University of Chicago.NMR relaxivity measurement
A high throughput MRI screening technique was used to evaluate the NMR relaxation properties of the HMN solutions at 3 Tesla field strength using a method similar to that previously reported.45 For MRI relaxivity measurements, formulations of SPION loaded mesophase nanoparticle and aqueous solutions of phase transferred SPIONs were placed in a multiwell plate at five different Fe concentrations serially diluted from 200 μM to 12.5 μM.The multiwell plate loaded with sample solutions was then imaged at 23 °C in a Siemens (Germany) 3 Tesla TRIO MRI scanner using a body transmit radio frequency coil and a 12 channel radio frequency receiver coil. The procedure for the quantification of transverse relaxation rate constant R2 has been described elsewhere.12 The transverse relaxivity, r2, of the HMNs is the linear gradient of the R2 data plotted as a function of Fe concentration. A linear least squares analysis (Matlab®) was used to quantify the transverse relaxivity.
Operative procedure for animal MRI tests
Approval for this study was obtained from the Florey Neuroscience Institutes animal ethics committee (AEC No. 06–073). In a typical experiment, an adult male Sprague-Dawley rat weighing 291 g was anesthetized with 5% isoflurane in a 1 : 1 mixture of medical grade air and oxygen and prepared for surgery. Anesthesia was maintained throughout the procedure and subsequent imaging with 1–2.5% isoflurane through a nose-cone placed over the animal's snout. The femoral vein was exposed, cannulated, and the incision closed with silk sutures. Anesthesia was maintained as the animal was prepared for magnetic resonance imaging (MRI).Animal MRI measurements
The anesthetized animal was laid supinely on an animal holder with respiration continuously monitored throughout the experiment with a pressure sensitive probe positioned over the rat's diaphragm. The cradle was inserted into a transmit/receive coil fixed inside a BGA12S gradient set for imaging with a 4.7 Tesla Bruker Biospec 47/30 scanner. The scanning protocol consisted of a 3-plane localizer sequence followed by multi-slice axial, coronal and sagittal scout images to ascertain the orientation and position of the kidneys and liver. Two oblique slices were selected; the first orientated to image the midline of both kidneys while the second dissected the liver. T2-weighted images were acquired prior to, and after, injection of contrast enhancing HMNs using a rapid acquisition, relaxation enhanced (RARE) sequence with the following imaging parameters: recovery time (TR) = 2930.25 ms, RARE factor = 8, effective echo time (TEeff) = 38.874 ms, field of view (FOV) = 6 cm2, matrix size = 256 × 256, in-plane resolution = 234 μm2, number of slices = 2, slice thickness = 4 mm and averages (NEX) = 4, for a total scan time of 3 min 12 s.Intravenous administration of contrast agent to animals
After acquiring a baseline scan, 1 mL of filtered (0.45 μm) HMNs containing 8 nm SPIONs (SPION loading at 0.3 wt% of Fe in phytantriol) in phosphate buffer at an iron concentration of 0.7 mM, a phytantriol concentration of 38 mM and an F-127 concentration of 0.7 mM were injected intravenously followed by approximately 0.3 ml of saline to ensure the cannula line was completely flushed of contrast agent. The total injection was performed slowly over a period of approximately 60 s and the animal's respiration rate was continuously monitored. The post injection scan was acquired immediately after administration of the contrast agent and then every five minutes thereafter for half an hour.Results and Discussion
The iron oxide nanoparticles used in this work for incorporation into the phytantriol cubic phase nanoparticles were spherical and highly monodispersed with an average diameter of 8 ± 0.04 nm as observed by cryo-TEM (Fig. 1). Details of the physical properties, structures and magnetic properties of the SPIONs were reported elsewhere.46 In brief, X-ray diffraction measurements showed that the nanoparticles were composed of magnetite (Fe3O4) and, more likely, maghemite (γ-Fe2O3). The nanoparticles are polycrystalline in nature with a crystallite size of 2.8 nm. Magnetic properties measurements showed that a near saturation magnetization value (Ms) of 31 emu g−1 was obtained at a magnetic field above 40000 Oe and the magnetic hysteresis was close to zero.
 | ||
| Fig. 1 Transmission electron microscopy image of hydrophobic oleic acid capped SPIONs with an average particle size of 8 nm. | ||
Dispersions of the cubic phase nanoparticles loaded with magnetic nanoparticles and stabilized by the pluronic F-127 block copolymer were prepared by dispersion in water at high temperature using homogenization. The light brown solutions of hybrid mesophase nanoparticles (HMNs) were freely flowing, with an apparent viscosity comparable to that of water. The solutions were stable for at least 6 months at room temperature without any significant changes in appearance and able to be filtered through a 0.45 μm filter. Particle sizes of the HMNs were determined by dynamic light scattering (DLS) and the intensity versus particle size distribution profile of a typical formulation is shown in Fig. 2. The DLS results for all HMNs produced showed a monomodal particle size distribution, with peaks centred around 180–200 nm. A similar average particle size and size distribution pattern was observed for cubic phase nanoparticles without SPIONs (data not shown). No significant change in particle size distribution was observed when the samples were analysed after several months.
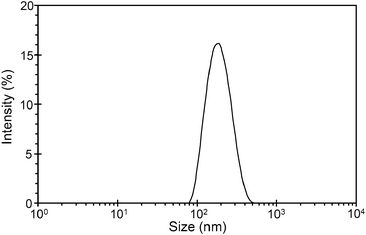 | ||
| Fig. 2 Intensity-weighted average particle size distribution of SPION-loaded cubic phase nanoparticles (HMNs). | ||
Cryo-TEM was used to examine the HMNs loaded with 8 nm SPIONs and the images are shown in Fig. 3. These images show the presence of discrete colloidal particles over a size range of between 60 nm and 300 nm which are typical of these systems and are consistent with the size obtained from DLS experiments. The images clearly show nanostructured particles displaying a periodic internal structure typical of cubic phase materials. The HMNs displayed various shapes and sizes ranging from square edged, to polyhedral and essentially spherical in shape, depending on the orientation of these particles with respect to the plane of observation. The observed patterns within the particles, which are typical for cubic phase nanoparticles also depend on the viewing angles.47,48 Vesicular dispersions are also seen in the images as spheres without any internal structures. Some of the HMNs appear to be fused and the distribution of SPIONs within the HMNs appears to be quite inhomogeneous. In the images, SPIONs are seen as black dots, more often within a cluster and are accompanied by local ordering inside the bulk ordered structure of the lipid aggregate probably due to compatibility of the hydrophobic chain (oleat moiety) on the surface of SPIONs. Close examination of the cryo-TEM images does not indicate any visible disturbance in the local structure of the bulk cubic phase lipid due to the presence of the nanoparticles. It is evident from the images that the SPIONs are not distributed uniformly within the mesophase particles and not all particles contain SPIONs. This is most likely due to inhomogeneity of the distribution of SPIONs in phytantriol.
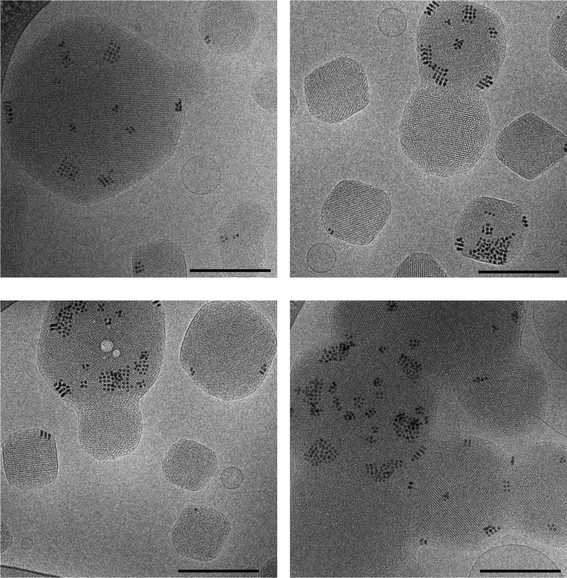 | ||
| Fig. 3 Cryo-TEM images of mesophase nanoparticles loaded with 8 nm spherical SPIONs at a concentrations of 0.3 wt% Fe in phytantriol. The scale bar corresponds to 200 nm. Ordering of the SPIONs within the bulk cubic phase nanoparticle materials can be seen. | ||
HMNs were analyzed by synchrotron SAXS and the results for the SPION-loaded cubic nanoparticle system (at a loading of 0.3 wt% of Fe in Phytantriol) are shown in Fig. 4 as an example. The observed SAXS pattern is the contribution of both SPIONs and the bicontinuous cubic phase. There is a hump/band in the low q region which can be attributed to the scattering from SPIONs... Six peaks observed in the region of q = 0.10–0.30 A−1 (indicated by arrows in Fig. 4) arise from the internal periodic structure of the bicontinuous cubic phase of phytantriol. These diffraction peaks which can be indexed as hkl = 110, 111, 200, 211, 220, 221/300 correspond to the Q224 (diamond) structure, that is, the cubic phase having Pn3m symmetry with peak positions at √2, √3, √4, √6, √8, √9 and a lattice parameter, a, given by:
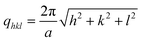 | (1) |
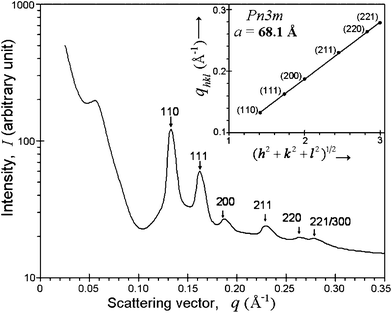 | ||
| Fig. 4 SAXS pattern of 8 nm SPION-loaded cubic phase nanoparticles in phytantriol at a concentration of 0.3 wt% of Fe in phytantriol. The peaks indicated by arrows (indexed as hkl = 110, 111, 200, 211, 220, 221/300 from left to right) confirm a Pn3m structure (Q224 phase). The lattice parameter (a) of 68.1 Å is obtained from the gradient of the plot of qhkl as a function of (h2+k2+l2)1/2 for these peaks (shown in inset) using eqn (1). | ||
To assess the effect of increasing SPION concentration within the phytantriol HMNs on T2 relaxivity (r2), an experiment was performed in which different formulations of cubic phase nanoparticles were prepared with increasing concentrations of SPIONs and the transverse relaxivity of each of these formulations was measured. Plots of transverse relaxivity (r2) of the SPIONs inside the cubic phase nanoparticles as a function of wt% of Fe in the bulk phytantriol of the HMNs are shown in Fig. 5. It can be seen that there is a linear trend with increasing relaxivity with increasing concentration of SPION in the HMNs. The concentrations of SPION in the HMNs were kept below 0.3 wt% as above this amount the precipitation of undispersed SPIONs was noticeable after homogenization, particularly after several weeks of storage. This indicated an upper limit to the amount of SPIONs that can be added to the bulk cubic phase of the phytantriol nanoparticles. The relaxivity of the monodispersed 8 nm SPIONs when using a polymer stabiliser was ca. 17.3 mM−1 s−1 which is significantly smaller than that of a previously clinically used product Resovist® which has a T2 relaxivity of ca. 207 mM−1 s−1 when measured under the same conditions.46 It should be noted that Resovist® consists of groups of agglomerated SPIONs inside a dextran polymer matrix. This agglomeration of SPIONs in Resovist® results in its higher relaxivity when compared to our monodispersed SPIONs of a similar size used in this work. At low loadings of SPION in the HMNs there was a significant enhancement (more than a doubling) in the T2 relaxivity of the HMNs compared to monodispersed suspensions of SPIONs phase transferred using a maleic anhydride copolymer.46 An essentially linear increase in the HMNs relaxivities with increasing loading of SPIONs was observed and the relaxivity value of ca. 80 mM−1 s−1 was observed at the highest loading of SPIONs tested. As the magnetic nanoparticles were not expected to be in direct contact with water, the enhanced relaxivities of the HMNs indicate that the magnetic moments of the SPIONs are able to interact with water inside the bi-continuous water channels and the surrounding bulk water of the dispersed, discrete HMNs and hence effectively reduce the transverse relaxation times.
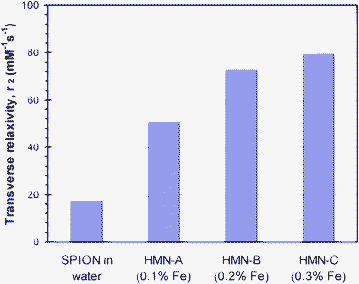 | ||
| Fig. 5 Plot of T2 relaxivity (mM−1 s−1), r2, of 8 nm SPION in water and in cubic mesophase nanoparticles (HMNs) at different loadings of 8 nm SPIONs in the HMNs in solutions labelled as HMN-A, HMN-B and HMN-C. SPIONs loadings increase from 0.1 to 0.3 wt% of Fe in phytantriol respectively and the concentration of phytantriol in the HMN solutions is 5.5 wt%. Resovist® has a T2 relaxivity value of 206.96 mM−1 s−1 measured under same conditions (from Ref. 46). | ||
The reason for the enhanced relaxivities generated from the dispersing of SPIONs in the phytantriol nanoparticles can be attributed to a number of physical phenomena. Firstly, the HMNs presumably tumble in solution at a slower rate than monodisperse SPIONs. Therefore, rotational correlation effects could be expected to play a role in enhancing the relaxivities observed when the particles are dispersed in the HMNs. Secondly, the observed trend may be due in part to aggregation and ordering of the SPIOs inside the bulk cubic phase structure of the HMNs. Greater inter-SPION distances are expected within the bulk mesophase at lower loadings and minimal influence of the magnetization of one particular SPION to an adjacent particle is anticipated. The transverse relaxation time, T2, depends, in part, on the relationship between the diffusion time of water molecules moving through the induced magnetic field fluctuations of the SPIONs and the pulse echo time. Hence, for the same matrix and experimental conditions, only marginal effects on the relaxivity would be expected with increasing concentration or number density of SPIONs in the HMNs if there is no interaction between the magnetic moments of individual SPIONs in the HMNs.49,50 However, a strong enhancement in T2 relaxivity has been observed when magnetic nanoparticles are within close proximity or clustered.51–53 In addition the shape and density of clusters significantly enhances the relaxivity behavior of SPIONs.51–54 The mechanism of the interaction is complex and not fully elucidated, however Monte Carlo simulations have shown that the proximity of nanoparticles within a cluster, their shape, size and polydispersity affect the magnetization processes.55–57 Another possible reason for the increase in relaxivity observed with increasing SPION concentration may be due to the inhomogeneous distribution of SPIONs within the bulk mesophase and variations in the cluster sizes and shapes. This would lead to magnetization differences in discrete regions in the HMNs which may give rise to local field gradients that accelerate the loss of phase coherence of the spins contributing to the MR signal producing an enhancement of r2.
Although encapsulation of SPIONs within the cubic mesophase improved their T2-relaxivity value, the relaxivity of HMNs at the highest loading of SPIONs is lower than that of Resovist®. However, it should be noted that although the absolute relaxivity is one of the important parameters used to evaluate the clinical potential of contrast agents, there are other factors such as the shelf life stability of the material, in vivo performance, biodistribution, cytotoxicity, and length of the subsequent imaging window (pharmacokinetics) must be taken into account when considering a new contrast agents suitability for use and further development.
We proposed in this work that the HMNs studied may have the potential to be used as MRI contrast agents by the shortening of T2 relaxation times. It was envisioned that subsequent to bolus delivery of HMNs, the reticuloendothelial system (RES) would transport the HMNs to hepatic and renal organs. Clinically approved superparamagnetic iron oxide nanoparticles (SPIONs) tend to be found in the liver after injection due to the RES, and have been extensively used for diagnosis of small lesions, metastasis, fibrosis and cirrhosis of the liver. In this work it was envisioned that the HMNs would perform similarly to SPION formulations such as Resovist® (a liver contrast agent) hence T2 weighted MR scans of the liver and kidneys were conducted both prior and subsequent to, intravenous injection of 1 mL of 8 nm SPIO containing HMNs with a relaxivity of ca. 80 mM−1 s−1 in phosphate buffer. The injected solution was at a concentration of 0.7 mM with respect to Fe, 38 mM with respect to phytantriol and 0.7 mM with respect to the pluronic polymer stabiliser, F-127. Excellent contrast in T2 weighted magnetic resonance images was observed in the liver and kidneys of live rats after injection of the HMNs. The signal decrease in the liver was normalized to the surrounding mean muscle tissue signal and false colour images are reported in Fig. 6. T2 weighted signal loss was observed in the liver and kidneys of animals shortly following injection of the HMNs.
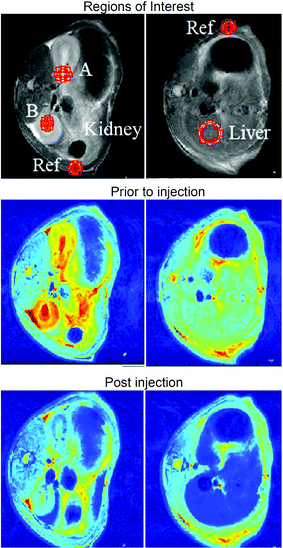 | ||
| Fig. 6 False colour T2 weighted MR images of the kidney (left column) and liver (right column) prior to, and after, intravenous injection of 1 mL of 8 nm HMNs with a relaxivity of ca. 80 mM−1 s−1 in phosphate buffer (left and right, respectively). The injected solution was at a concentration of 0.7 mM wrt Fe, 38 mM wrt phytantriol and 0.7 mM wrt F-127. | ||
When the animals were scanned up to one hour post injection the same level of contrast was observed in the liver and a slightly reduced signal was observed in the kidneys (data not shown). We did not observe contrasts in other organs. Our ethics approval did not allow us to conduct a study beyond this time point and the animals were sacrificed after one hour of anaesthetisation. Resovist® has been used in research labs in animals (including rats) at similar boluses and similar contrast in the liver and kidneys is observed for Resovist® as we have seen.39,58 However, Resovist® has been withdrawn from the market and is no longer available for clinical use in Australia and America.
A significant challenge in the field of nanoparticulate based MRI contrast and drug delivery agents is the need to target tissues and organs of interest such as cancerous lesions in vivo and prolong blood half life. In order to fulfil this goal such nanoparticles must be protected from sequestration by the RES. This is a significant challenge. To date we have shown that the blood half-life of mesophase nanoparticles in rats can be improved by an order of magnitude when a PEG lipid is used in conjunction with F-127 as a stabilizer.32 Ongoing work will be required to further improve the pharmacokinetcs of this class of lipidic mesophase nanoparticle in future studies. The results of this study show that HMNs can effectively produce negative contrast in T2 weighted magnetic resonance images in the kidneys and liver of live rats and have potential as T2 MRI contrast agents.
Conclusions
In summary, colloidally stable, T2 enhancing SPION containing lyotropic liquid crystal hybrid nanoparticles (HMNs) have been produced that provide effective in vivo MRI contrast enhancement in the liver and kidneys or rats. Incorporation of an 8 nm SPION into the cubic mesophase liquid crystal nanoparticle structure allows for a significant enhancement of relaxivity compared to monodisperse SPIONs in solution which we attribute to rotational correlation constant effects and greater ordering of the SPIONs in the HMNs. The HMNs are tolerated in live animals and good contrast in the liver and kidneys of live rats is observed. These lyotropic liquid crystal nanoparticles have the potential to be multifunctional via incorporation of targeting, imaging and therapeutic agents.Acknowledgements
The authors would like to acknowledge the help of Nigel Kirby at SAXS/WAXS beamline at the Australian Synchrotron, Victoria, Australia with SAXS analysis. Authors are thankful to Guoliang Zhen and Paul Mulvalney for supplying iron oxide nanoparticles. GFE is a NHMRC Principal Research Fellow (#1003993). BAM was supported by an NHMRC Fellowship (#454790).References
- C. Corot, P. Robert, J. M. Idee and M. Port, Adv. Drug Delivery Rev., 2006, 58, 1471–1504 CrossRef CAS.
- A. Bjornerud and L. Johansson, NMR Biomed., 2004, 17, 465–477 CrossRef.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS.
- G. P. Krestin, Y. X. J. Wang and S. M. Hussain, Eur. Radiol., 2001, 11, 2319–2331 CrossRef.
- E. Canet, D. Revel, R. Forrat, C. Baldyporcher, M. Delorgeril, L. Sebbag, J. P. Vallee, D. Didier and M. Amiel, Magn. Reson. Imaging, 1993, 11, 1139–1145 CrossRef CAS.
- C. Chambon, O. Clement, A. Leblanche, E. Schoumanclaeys and G. Frija, Magn. Reson. Imaging, 1993, 11, 509–519 CrossRef CAS.
- Z. R. Stephen, F. M. Kievit and M. Q. Zhang, Mater. Today, 2011, 14, 330–338 CrossRef CAS.
- H. Yang, U. Jeong, X. W. Teng, Y. Wang and Y. N. Xia, Adv. Mater., 2007, 19, 33–60 CrossRef.
- B. A. Moffat, G. R. Reddy, P. McConville, D. E. Hall, T. L. Chenevert, R. R. Kopelman, M. Philbert, R. Weissleder, A. Rehemtulla and B. D. Ross, Mol. Imaging, 2003, 2, 324–332 CrossRef CAS.
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS.
- P. Reimer, B. Tombach, H. Daldrup, T. Hesse, G. Sander, T. Balzer, K. Shamsi, T. Berns, E. J. Rummeny and P. E. Peters, Radiologe, 1996, 36, 124–133 CrossRef CAS.
- B. W. Muir, B. A. Moffat, P. Harbour, G. Coia, G. L. Zhen, L. Waddington, J. Scoble, D. Krah, S. H. Thang, Y. K. Chong, P. Mulvaney and P. Hartley, J. Phys. Chem. C, 2009, 113, 16615–16624 CAS.
- E. E. Lees, T. L. Nguyen, A. H. A. Clayton, P. Mulvaney and B. W. Muir, ACS Nano, 2009, 3, 1121–1128 CrossRef CAS.
- E. Amstad, S. Zurcher, A. Mashaghi, J. Y. Wong, M. Textor and E. Reimhult, Small, 2009, 5, 1334–1342 CrossRef CAS.
- M. Meincke, T. Schlorf, E. Kossel, O. Jansen, C.-C. Glueer and R. Mentlein, Front. Biosci., 2008, 13, 4002–4008 CrossRef CAS.
- C. Faure, M. E. Meyre, S. Trepout, O. Lambert and E. Lebraud, J. Phys. Chem. B, 2009, 113, 8552–8559 CrossRef CAS.
- J. A. Dagata, N. Farkas, C. L. Dennis, R. D. Shull, V. A. Hackley, C. Yang, K. F. Pirollo and E. H. Chang, Nanotechnology, 2008, 19, 305101 CrossRef CAS.
- D. Constantin, P. Davidson and C. Chaneac, Langmuir, 2010, 26, 4586–4589 CrossRef CAS.
- S. B. Lecommandoux, O. Sandre, F. Checot, J. Rodriguez-Hernandez and R. Perzynski, Adv. Mater., 2005, 17, 712–718 CrossRef CAS.
-
P. Hartley and A. Polyzos, PCT Int. Appl., WO 2010060131 A1, 2009.
- K. Larsson, J. Phys. Chem., 1989, 93, 7304–7314 CrossRef CAS.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2000, 5, 64–69 CrossRef CAS.
- K. Larsson, J. Dispersion Sci. Technol., 1999, 20, 27–34 CrossRef CAS.
- J. Gustafsson, H. LjusbergWahren, M. Almgren and K. Larsson, Langmuir, 1996, 12, 4611–4613 CrossRef CAS.
- C. Guo, J. Wang, F. Cao, R. J. Lee and G. Zhai, Drug Discovery Today, 2010, 15, 1032–1040 CrossRef CAS.
- L. Sagalowicz, M. E. Leser, H. J. Watzke and M. Michel, Trends Food Sci. Technol., 2006, 17, 204–214 CrossRef CAS.
- B. J. Boyd, Y. D. Dong, I. Larson and T. Hanley, Langmuir, 2006, 22, 9512–9518 CrossRef.
- A. Yaghmur and O. Glatter, Adv. Colloid Interface Sci., 2009, 147–148, 333–342 CrossRef CAS.
- N. Garti, I. Amar-Yuli, D. Libster and A. Aserin, Curr. Opin. Colloid Interface Sci., 2009, 14, 21–32 CrossRef.
- C. Y. Guo, J. Wang, F. L. Cao, R. J. Lee and G. X. Zhai, Drug Discovery Today, 2010, 15, 1032–1040 CrossRef CAS.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS.
- B. W. Muir, D. P. Acharya, D. F. Kennedy, X. Mulet, R. A. Evans, S. M. Pereira, K. L. Wark, B. J. Boyd, T.-H. Nguyen, T. M. Hinton, L. J. Waddington, N. Kirby, D. K. Wright, H. X. Wang, G. F. Egan and B. A. Moffat, Biomaterials, 2012, 33, 2723–2733 CrossRef CAS.
- S. J. Fraser, R. M. Dawson, L. J. Waddington, B. W. Muir, X. Mulet, P. G. Hartley, F. Separovic and A. Polyzos, Aust. J. Chem., 2011, 64, 46–53 CrossRef CAS.
- S. M. S. S. M. Sagnella, X. J. Gong, M. J. Moghaddam, C. E. Conn, K. Kimpton, L. J. Waddington, I. Krodkiewska and C. J. Drummond, Nanoscale, 2011, 3, 919–924 RSC.
- X. Mulet, D. F. Kennedy, C. E. Conn, A. Hawley and C. J. Drummond, Int. J. Pharm., 2010, 395, 290–297 CrossRef CAS.
- S. J. Fraser, X. Mulet, L. Martin, S. Praporski, A. Mechler, P. G. Hartley, A. Polyzos and F. Separovic, Langmuir, 2012, 28, 620–627 CrossRef CAS.
- S. J. Fraser, R. Rose, M. K. Hattarki, P. G. Hartley, O. Dolezal, R. M. Dawson, F. Separovic and A. Polyzos, Soft Matter, 2011, 7, 6125–6134 RSC.
- W. K. Fong, T. L. Hanley, B. Thierry, N. Kirby and B. J. Boyd, Langmuir, 2010, 26, 6136–6139 CrossRef CAS.
- L. Kaufner, R. Cartier, R. Wustneck, I. Fichtner, S. Pietschmann, H. Bruhn, D. Schutt, A. F. Thunemann and U. Pison, Nanotechnology, 2007, 18, 115710 CrossRef.
- J. F. Lutz, S. Stiller, A. Hoth, L. Kaufner, U. Pison and R. Cartier, Biomacromolecules, 2006, 7, 3132–3138 CrossRef CAS.
- A. Hofmann, S. Thierbach, A. Semisch, A. Hartwig, M. Taupitz, E. Ruhl and C. Graf, J. Mater. Chem., 2010, 20, 7842–7853 RSC.
- S. W. Provencher, Makromol. Chem., 1979, 180, 201–209 CrossRef CAS.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 229–242 CrossRef.
- S. W. Provencher, J. Hendrix, L. Demaeyer and N. Paulussen, J. Chem. Phys., 1978, 69, 4273–4276 CrossRef CAS.
- D. Hogemann, V. Ntziachristos, L. Josephson and R. Weissleder, Bioconjugate Chem., 2002, 13, 116–121 CrossRef.
- G. L. Zhen, B. W. Muir, B. A. Moffat, P. Harbour, K. S. Murray, B. Moubaraki, K. Suzuki, I. Madsen, N. Agron-Olshina, L. Waddington, P. Mulvaney and P. G. Hartley, J. Phys. Chem. C, 2011, 115, 327–334 CAS.
- Z. A. Almsherqi, S. D. Kohlwein and Y. Deng, J. Cell Biol., 2006, 173, 839–844 CrossRef CAS.
- Z. A. Almsherqi, T. Landh, S. D. Kohlwein and Y. R. Deng, in International Review of Cell and Molecular Biology, Vol 274, ed. K. W. Jeon, 2009, pp. 275–342. Search PubMed.
- R. M. Westervelt, K. A. Brown, C. C. Vassiliou, D. Issadore, J. Berezovsky and M. J. Cima, J. Magn. Magn. Mater., 2010, 322, 3122–3126 CrossRef.
- R. A. Brooks, F. Moiny and P. Gillis, Magn. Reson. Med., 2001, 45, 1014–1020 CrossRef CAS.
- H. Ai, C. Flask, B. Weinberg, X. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. M. Gao, Adv. Mater., 2005, 17, 1949–1952 CrossRef CAS.
- D. X. Chen, N. Sun, Z. J. Huang, C. M. Cheng, H. Xu and H. C. Gu, J. Magn. Magn. Mater., 2010, 322, 548–556 CrossRef CAS.
- C. Paquet, H. W. de Haan, D. M. Leek, H. Y. Lin, B. Xiang, G. H. Tian, A. Kell and B. Simard, ACS Nano, 2011, 5, 3104–3112 CrossRef CAS.
- Y. Matsumoto and A. Jasanoff, Magn. Reson. Imaging, 2008, 26, 994–998 CrossRef CAS.
- V. Schaller, G. Wahnstrom, A. Sanz-Velasco, P. Enoksson and C. Johansson, J. Magn. Magn. Mater., 2009, 321, 1400–1403 CrossRef CAS.
- V. Schaller, G. Wahnstrom, A. Sanz-Velasco, P. Enoksson and C. Johansson, International Conference on Magnetism (ICM 2009), 2010, vol. 200, 072085 Search PubMed.
- V. Schaller, G. Wahnstrom, A. Sanz-Velasco, S. Gustafsson, E. Olsson, P. Enoksson and C. Johansson, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 092406 CrossRef.
- J.-C. Jao, H.-Y. Lu, H.-C. Lu, G.-C. Liu, S.-H. Chen, S.-L. Lian, S.-F. Yang and P.-C. Chen, 2010 4th International Conference on Bioinformatics and Biomedical Engineering (iCBBE), IEEE Conference Publication, 2010, DOI:10.1109/ICBBE.2010.5517191..
| This journal is © The Royal Society of Chemistry 2012 |