Coal fly ash supported Co3O4 catalysts for phenol degradation using peroxymonosulfate
Syaifullah
Muhammad
ab,
Edy
Saputra
ac,
Hongqi
Sun
a,
Juliana de C.
Izidoro
ad,
Denise Alves
Fungaro
d,
Ha Ming
Ang
a,
Moses O.
Tadé
a and
Shaobin
Wang
*a
aDepartment of Chemical Engineering, Curtin University, GPO Box U1987, WA 6845. E-mail: Shaobin.wang@curtin.edu.au; Fax: (+61)8 9266 2681; Tel: (+61)8 9266 3776
bDepartment of Chemical Engineering, Syiah Kuala University, Banda Aceh, Indonesia
cDepartment of Chemical Engineering, Riau University, Pakanbaru, Indonesia
dChemical and Environmental Technology Center, Nuclear and Energy Research Institute, P.C. 11049, São Paulo, Brazil
First published on 17th May 2012
Abstract
Several fly ash (FA) samples derived from Australian (FA-WA) and Brazilian coals (FA-JL and FA-CH) were used as supports to prepare Co oxide (Co)-based catalysts. These Co/FA catalysts were tested in peroxymonosulfate activation for sulphate radical generation and phenol degradation in aqueous solution. The physicochemical properties of FA supports and Co/FA catalysts were characterised by N2 adsorption, X-ray diffraction (XRD), scanning electron microscopy coupling with energy dispersive spectroscopy (SEM-EDS), elemental mapping, and UV-vis diffuse reflectance spectroscopy. It was found that the FA supports did not show adsorption of phenol and could not activate peroxymonosulfate for sulphate radical generation. However, fly ash supported Co oxide catalysts (Co/FA) presented higher activities in the activation of peroxymonosulfate for phenol degradation than bulk Co oxide and their activities varied depending on the properties of the fly ash supports. Co/FA-JL showed the highest activity while Co/FA-WA showed the lowest. Activation energies of phenol degradation on three Co/FA catalysts were obtained to be 47.0, 56.5, 56.0 kJ mol−1 for Co/FA-WA, Co/FA-JL and Co/FA-CH, respectively.
Introduction
Currently, large amounts of solid waste is produced from various industries. Fly ash from coal, oil and biomass combustion is one of the major contributors to solid waste and the current practice is disposal in landfills or dumping at sea. In the past years, fly ash has been explored for several applications in adsorption,1–3 material synthesis such as zeolites,4–6 geopolymers,7–9 ceramics,10,11 and catalyst supports.12–14Wastewater contains many different pollutants, such as dusts, metal ions, and organic compounds. Removal of organic pollutants is an important process in water and wastewater treatments. The processes for organic removal include adsorption, flocculation, membrane separation and oxidation. In the last few decades, advanced oxidation processes (AOPs) have emerged as effective techniques to degrade organic compounds for wastewater treatment. Currently, most of AOPs are based on the generation of very reactive species, such as hydroxyl radicals (OH•) that can oxidise a broad range of pollutants quickly and non selectively.15–17 Apart from OH•, sulphate radicals have been attracting high interest and proposed as an alternative due to their higher oxidation potential.18
Sulfate radicals can be generated from two oxidants, persulfate and peroxymonosulfate (PMS). For the activation of persulfate and peroxymonosulfate, metal ions are generally used, as shown in the following equations.19,20
Mn+ + HSO5−→ M(n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +1)+ + SO4•− + OH− +1)+ + SO4•− + OH− | (1) |
| Mn+ + S2O82− → M(n+1)+ + SO4•− + SO42− | (2) |
It has been found that homogeneous Co2+ is the best metal ion for the activation of peroxymonosulfate in order to produce sulfate radicals. However, Co2+ could cause environmental problems if it was present in water. Heterogeneous activation of peroxymonosulfate would provide a good solution. In the past few years, several supported Co systems have been investigated and show high activity.21–28
Resource recovery is one of the most effective strategies in waste management. Using solid waste for other applications provides a route for solid waste recycling and a reduction in waste disposal to landfills, bringing in environmental benefits and economic profits. In this paper, we report an investigation of the preparation of Co oxide catalysts on different sources of fly ash (FA) samples. We will study the effects of fly ash structure and the property on the Co/FA catalysts in activation of peroxymonosulfate for sulfate radical production in phenol degradation.
Experimental section
Materials and catalyst preparation
Fly ash samples were obtained from a coal-fired power station in Western Australia (WA) and two coal-fired power stations, Charqueadas Power Plant (CH) and Jorge Lacerda Power Plant (JL), Brazil. These fly ash samples were labelled as FA-WA, FA-CH, and FA-JL, respectively. Phenol, cobalt nitrate (Co(NO3)2·6H2O), and oxone (2KHSO5·KHSO4·K2SO4) were obtained from Sigma-Aldrich. A stock solution of phenol at 1000 ppm was prepared using deionised water.Cobalt oxide (Co3O4) was obtained by thermal decomposition of Co(NO3)2 at 500 °C for 2 h. For the synthesis of Co loaded catalysts, an impregnation method was used. Typically, 1.23 g of Co(NO3)2·6H2O was dissolved in 100 mL of ultrapure water. Then 5 g of FAs were added followed by stirring continuously at 80 °C until total evaporation of the H2O occurred. Furthermore, the sample was dried at 120 °C overnight and calcined at 500 °C for 4 h in air. Then the catalyst was stored in a desiccator until use. In the Co/FA samples, Co loading was kept at 5 wt%.
Characterisation
The crystal structure of the FA and Co/FA powders was characterised by an X-ray diffractometer (XRD, Bruker D8 Advance) equipped with Cu-Kα radiation, at an accelerating voltage and current of 40 kV and 40 mA, respectively. The surface area was measured by N2 adsorption/desorption analyser (Quantachrome Nova – 1200). Prior to adsorption, samples were degassed under high vacuum at 150 °C for 12 h. The BET surface areas were obtained by applying the BET equation to the nitrogen adsorption data. For measurements of sample pH, fly ash samples (0.1 g) were placed in 10 mL of deionised water and the mixture was stirred for 24 h in a shaker at 120 rpm. After filtration, the pH of the solutions was measured with a pH meter (MSTecnopon – Mod MPA 210). In cation exchange capacity (CEC) measurements, the samples were saturated with sodium acetate solution (1 M), washed with distilled water (1 L) and then mixed with ammonium acetate solution (1 M). The sodium ion concentration of the resulting solution was determined by optical emission spectrometry with inductively coupled plasma – ICP-OES (Spectroflame - M120).The UV-visible diffuse reflectance spectra (DRS) were recorded on a V-570 UV-visible spectrometer (Jasco, Japan) equipped with an integrating sphere, in which BaSO4 was used as a reference material. Scanning electron microscopy (SEM), performed on a Neon 40EsB FIBSEM (Zeiss, Germany), was used to evaluate the morphology, size and textural information of the samples. The integrated energy dispersive spectroscopy (EDS) and elemental mapping (cobalt) were applied to analyse the dispersion of cobalt in the Co/FA samples.
Catalytic evaluation
Phenol degradation tests were carried out at 25–45 °C in a 1 L glass vessel with 500 mL of phenol solution at varying concentrations (ppm) with a constant stirring of 400 rpm. Firstly, 0.20 g of catalyst was added into the phenol solution for a while, then oxone was added into the solution at 2 g L−1, unless otherwise indicated. At a certain time, a water sample (1 mL) was withdrawn into a HPLC vial and 0.5 mL of pure methanol was injected into the vial to quench the reaction. The concentration of phenol was analysed using a Varian HPLC with a UV detector set at λ = 270 nm. A C–18 column was used to separate the organics while the mobile phase with a flowrate of 1.5 mL min−1 was made of 30% CH3CN and 70% water. For selected samples, total organic carbon (TOC) was obtained using a Shimadzu TOC-5000 CE analyser.Results and discussion
Characterisation of FA supports and Co/FA catalysts
The chemical compositions of three fly ash samples (by weight%) are given in Table 1. Fly ash is mainly composed of metal oxides derived from inorganic compounds in coals. Major constituents for all fly ash samples are: SiO2, Al2O3, and Fe2O3. The contents of SiO2 and Al2O3 are above 70% for all samples. SiO2/Al2O3 for the three samples is in the order of FA-WA > FA-CH > FA-JL. Higher SiO2/Al2O3 concentrations could result in a lower pH of solids due to the acidity of SiO2. A comparison of three fly ashes shows that FA-CH has the highest SiO2 and Al2O3 contents but the lowest content of Fe2O3. FA-JL has the medium contents of Al2O3 and Fe2O3, but it has the highest contents of K2O, CaO, and MgO. FA-WA has the highest content of Fe2O3 but low contents of K2O, CaO, and MgO.| Components | FA-WA | FA-JL | FA-CH |
|---|---|---|---|
| SiO2 | 55.0 | 50.3 | 57.5 |
| Al2O3 | 29.3 | 29.8 | 32.6 |
| Fe2O3 | 8.8 | 6.70 | 3.60 |
| K2O | 0.4 | 5.30 | 2.00 |
| CaO | 1.6 | 2.70 | 1.40 |
| TiO2 | — | 2.20 | 1.60 |
| SO3 | 0.1 | 1.40 | 0.40 |
| MgO | 1.0 | 1.10 | 0.70 |
| Na2O | 0.3 | — | — |
| SiO2/Al2O3 | 1.88 | 1.69 | 1.76 |
The physicochemical properties of three fly ashes are presented in Table 2. FA-JL and FA-CH show a much high pH, larger than 7, while FA-WA shows a lower pH, less than 4. This suggests that FA-JL and FA-CH demonstrate strong basic surfaces and FA-WA presents an acidic surface. This is attributed to the different chemical compositions of the FA samples. Table 1 shows that FA-JL and FA-CH have higher contents of Al2O3, K2O, CaO and MgO, which are basic oxides, making them more strongly basic. FA-JL has the highest contents of K2O, CaO and MgO and thus has the highest pH. For the loss on ignition (LOI) and surface area, FA-JL and FA-WA have higher values than FA-CH. In general, LOI is the indicator of unburned carbon content. Unburned carbons are porous materials formed during high-temperature processing, which will gave FA a high surface area. For the three FA samples, the CEC is very similar.
| Sample | pH | Loss on ignition (LOI) (%) | S BET (m2 g−1) | CEC (meq g−1) |
|---|---|---|---|---|
| FA-JL | 8.0 | 15.1 | 9.6 | 0.026 |
| FA-CH | 7.8 | 2.60 | 3.3 | 0.026 |
| FA-WA | 3.7 | 5.2 | 15.6 | 0.029 |
Fig. 1 shows XRD patterns of the FA samples and their supported Co catalysts. Three fly ashes show very similar patterns and the crystalline phases were identified mainly as mullite, quartz and minors as hematite and magnetite, which are confirmed from their chemical compositions (Table 1). For Co/FA catalysts, Co3O4 peaks were identified on all samples, which is due to the decomposition of Co(NO3)2. It has been reported that Co(NO3)2 decomposition will produce CoO, Co2O3 and Co3O4 and that the final product is Co3O4. XRD results thus showed that only Co3O4 was presented on Co/FA catalysts.
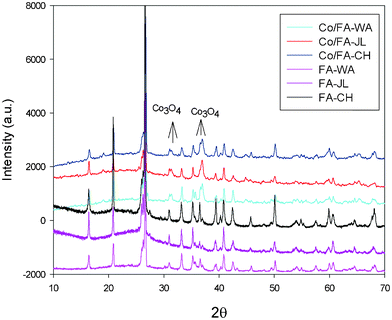 | ||
| Fig. 1 XRD patterns of fly ash and red mud and their supported Co catalysts. | ||
Fig. 2 shows SEM micrographs, EDS spectra and Co elemental mapping of three Co/FA catalysts. As shown, most particles of Co/FA-JL and Co/FA-WA catalysts presented as spherical particles while the particles of Co/FA-CH presented as irregular shape. EDS spectra of three catalysts showed the presence of C, O, Si, Al, Fe, Na, K, Ti, Mg, Ca, S, and Co on catalysts. Co loadings on three catalysts were derived as 5.4, 3.5 and 11.9 wt% on Co/FA-WA, Co/FA-CH and Co/FA-JL, respectively. Elemental mapping also showed the distribution of Co on the catalysts. Co on Co/FA-JL was much more homogeneously distributed on the surface. Co on Co/FA-CH also showed good dispersion but the intensity seemed lower than Co/FA-JL. However, Co on Co/FA-WA seemed to have some large black spots, which suggested that Co was not well distributed compared with the other two catalysts.
 | ||
| Fig. 2 SEM micrograph, EDS and Co elemental mapping of Co/FA catalysts. | ||
Fig. 3 shows UV-vis diffuse reflectance spectra of three Co/FA catalysts. One can see that Co/FA-JL and Co/FA-CH showed similar profiles and Co/FA-WA presented differently. For Co/FA-WA, two strong and broad bands centred at 400 and 700 nm were observed, which indicate the formation of Co3O4. The first band at ca. 400 nm can be assigned to the ligand-metal charge transfer (i.e., O2→Co2+), while the band at about 700 nm is corresponding to the O2−→Co3+ charge transfer.29,30 However, for Co/FA-JL and Co/FA-CH, the first broad band is centred at 460 nm, which suggests the presence of Co2+ in tetrahedral coordination.
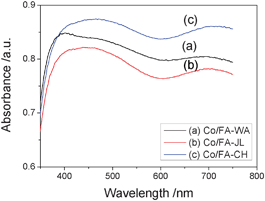 | ||
| Fig. 3 UV-vis diffuse reflectance spectra of Co/FA catalysts. | ||
Catalytic activity
Fig. 4 shows variation of phenol concentration with time for different solid systems in adsorption and catalytic oxidation. FA did not show strong phenol adsorption with about 5% reduction in phenol concentration at 90 min. For FA/oxone systems, phenol concentration showed a slightly higher reduction than that of adsorption with 10% phenol removal, suggesting that FA could slightly but not effectively activate oxone for sulfate radical production and phenol decomposition. The activation may be due to the presence of Fe oxide in the FA samples. Previous investigations have shown that Fe2+ could also activate PMS for sulfate radical generation, but the activity is quite low.19 Co3O4/oxone showed graduate phenol degradation, but phenol removal efficiency was close to that of FA/oxone. Unlike the FA and Co3O4 samples, the three FA supported Co catalysts showed much stronger phenol degradation, suggesting that dispersive Co oxides are capable of reacting with oxone for sulfate radical production. For Co/FA-WA, phenol concentration could be reduced by 40% after 90 min. Co/FA-CH showed better phenol reduction and achieved 56% phenol decomposition at 90 min. Co/FA-JL exhibited the best performance in phenol degradation with 70% phenol reduction at 90 min. The activity of the three catalysts followed the order of Co/FA-JL > Co/FA-CH > Co/FA-WA.![A comparison of phenol degradation on Co/FA catalysts. Reaction conditions: [Phenol] = 30 ppm, catalyst = 0.4 g L−1, oxone = 2 g L−1, T = 25 °C.](/image/article/2012/RA/c2ra20346d/c2ra20346d-f4.gif) | ||
| Fig. 4 A comparison of phenol degradation on Co/FA catalysts. Reaction conditions: [Phenol] = 30 ppm, catalyst = 0.4 g L−1, oxone = 2 g L−1, T = 25 °C. | ||
The difference in phenol degradation on three Co/FA catalysts is due to the varying surface properties of the catalysts. XRD and UV-vis diffuse reflectance spectra showed that Co3O4 is presented on three Co/FA samples, which will be the active species for activation of PMS. The heterogeneous activation of PMS can be expressed in following equations.
| S–Co(III) + HSO5− → S–Co(II) + SO5•− + H+ S−: support | (3) |
| S–Co(II) + HSO5− → S–Co(III) + SO4•− + OH− | (4) |
| C6H5OH + SO4•− → intermediates → CO2 + H2O + SO42− | (5) |
However, bulk Co3O4 did not show strong activity for phenol degradation while supported Co3O4 produced high phenol removal efficiency, suggesting the important role of Co3O4 dispersion. EDS and elemental mapping show that the dispersion of Co3O4 on FA supports is different. EDS and elemental mapping (Fig. 2) indicate Co/FA-JL has a higher Co dispersion, suggesting the presence of more active sites for PMS activation. UV-vis reflectance spectra showed the presence of Co2+ in tetrahedral coordination on Co/FA-JL and Co/FA-CH, which can lead to more SO4•− production (eqn (4)). In addition, Table 2 showed that FA-JL and FA-CH have basic surfaces and FA-WA has a strongly acid surface. The high surface basicity of FA-JL and FA-CH will promote the reaction with PMS. Zhang et al.24 investigated cobalt oxide catalysts immobilised on various oxides (MgO, ZnO, Al2O3, ZrO2, P25, SBA-15) for the degradation of organic dyes in dilute solutions with PMS and reported that Co/MgO catalysts were the most active. They suggested that the alkaline MgO support helped in (i) dispersing the cobalt oxide nanoparticles well, (ii) minimising the leaching of cobalt ions into the liquid phase, and (iii) facilitating the formation of surface Co–OH complex which is a critical step for PMS activation. Therefore, due to higher dispersion of Co3O4, Co2+ tetrahedral coordination, and strong basicity of FA-JL, Co/FA-JL exhibited the highest activity in phenol degradation. Liang et al.31 synthesised Al2O3−, SiO2− and TiO2− supported Co oxide catalysts and tested them for phenol degradation. They found that Co/Al2O3 exhibited higher activity than Co/SiO2. Table 1 shows the lower SiO2/Al2O3 content for FA-CH and FA-JL, which could result in higher activity of Co/FA-JL and Co/FA-CH.
Based on phenol degradation curves, a simple model, first order kinetics, was used to fit the data and produced good results (Table 3). The regression coefficients suggest that phenol degradation follows first-order kinetics. Some supported Co catalysts have been investigated for the activation of PMS in phenol degradation. Kinetic studies indicated that phenol degradation showed first order kinetics on Co/AC,23 similar to Co/FA catalysts in this investigation. The first-order kinetics suggests that phenol degradation occurred as surface reaction. HPLC analysis found the intermediates of hydroquinone and p-benzoquinone. Therefore, phenol degradation could be described in Scheme 1.
| Catalyst | First order kinetics | |
|---|---|---|
| K 1(min−1) | R | |
| Co/FA-WA | 0.0048±0.0004 | 0.980 |
| Co/FA-CH | 0.0086±0.0006 | 0.982 |
| Co/FA-JL | 0.0111±0.0006 | 0.990 |
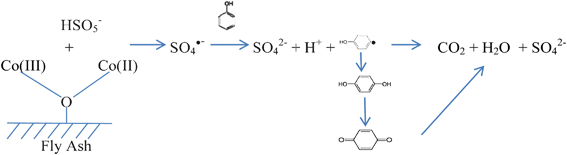 | ||
| Scheme 1 Co/FA activation of PMS for phenol degradation. | ||
Fig. 5 displays the phenol degradation efficiency at 90 min with different oxone loading in solution. Oxone concentration would affect phenol degradation rate and efficiency. Higher oxone loading will increase phenol degradation. At 0.3 g oxone, phenol degradation was about 70% and it could reach 99% at 1 g oxone. Phenol degradation depends on the generation of sulphate radicals. More oxone in solution will produce more sulphate radicals, leading to high phenol reduction.
![Effect of oxone loading on phenol degradation. Reaction conditions: [Phenol] = 30 ppm, catalyst = 0.4 g L−1, T = 25 °C.](/image/article/2012/RA/c2ra20346d/c2ra20346d-f5.gif) | ||
| Fig. 5 Effect of oxone loading on phenol degradation. Reaction conditions: [Phenol] = 30 ppm, catalyst = 0.4 g L−1, T = 25 °C. | ||
Fig. 6 illustrates phenol degradation at varying initial phenol concentrations. As can be seen, higher initial phenol concentration will result in low phenol degradation efficiency. At 30 ppm, phenol degradation was 99% but it would reduce to 30% at 100 ppm phenol. As stated before, phenol degradation is dependent on sulphate radicals. At the same concentrations of catalyst and PMS, high amount of phenol in solution will require more time to achieve the same removal rate, thus lowering phenol degradation efficiency.
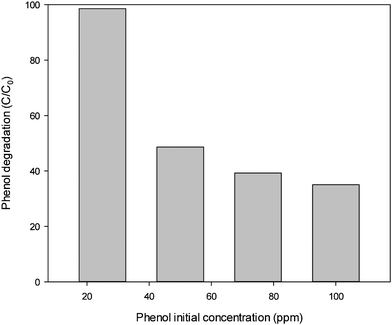 | ||
| Fig. 6 Effect of phenol initial concentration on phenol degradation. Reaction conditions: catalyst = 0.4 g L−1, oxone = 2 g L−1, T = 25 °C. | ||
Fig. 7 presents phenol degradation at varying temperatures on three Co/FA catalysts. In general, higher temperatures result in high phenol degradation for the three catalysts. At 45 °C, phenol degradation could achieve 100% at 90, 40 and 50 min on Co/FA-WA, Co/FA-JL, and Co/FA-CH, respectively. Based on the first order kinetics, the reaction rate constants were obtained for the three catalysts at different temperatures. A relationship between the rate constant and temperature could be described by the Arrhenius plots and activation energies were obtained (Table 4). The results indicated that Co/FA-JL, and Co/FA-CH presented similar activation energies but Co/FA-WA showed a lower value.
![Effect of temperature on phenol degradation on Co/FA catalysts. Reaction conditions: [Phenol] = 30 ppm, oxone = 2 g L−1, catalyst = 0.4 g L−1.](/image/article/2012/RA/c2ra20346d/c2ra20346d-f7.gif) | ||
| Fig. 7 Effect of temperature on phenol degradation on Co/FA catalysts. Reaction conditions: [Phenol] = 30 ppm, oxone = 2 g L−1, catalyst = 0.4 g L−1. | ||
| Catalysts | E a (kJ) | R 2 |
|---|---|---|
| Co/FA-WA | 47.0 | 0.993 |
| Co/FA-CH | 56.0 | 0.999 |
| Co/FA-JL | 56.5 | 0.999 |
For PMS activation by heterogeneous Co catalysts in phenol degradation, few investigations have reported the kinetics and activation energies. We have studied several heterogeneous Co catalysts on various supports in activation of PMS for phenol degradation. The activation energies obtained are 67.4,25 69.7,22 59.7 23 for Co/SBA-15, Co/ZSM5, and Co/AC, respectively. As seen that Co/FAs presented lower activation energy than activated carbon and oxide supported Co catalysts, suggesting that the Co/FA systems could be promising catalysts.
Conclusions
Three fly ash samples from different sources were used for the synthesis of supported Co catalysts. These catalysts showed higher activity than the supports and bulk Co3O4 in the activation of PMS for generation of sulphate radicals to oxidatively decompose phenol in aqueous solution. Co dispersion and support basicity strongly influence the activity of Co/FAs. Concentrations of oxone and phenol also influence phenol degradation efficiency. Kinetic studies showed phenol degradation followed a first order kinetics and the activation energies for Co/FA-WA, Co/FA-JL, and Co/FA-CH are 47.0, 56.5, 56.0 kJ mol−1, respectively.References
- S. B. Wang, Y. Boyjoo and A. Choueib, Chemosphere, 2005, 60, 1401–1407 CrossRef CAS.
- S. B. Wang, Y. Boyjoo, A. Choueib and Z. H. Zhu, Water Res., 2005, 39, 129–138 CrossRef CAS.
- S. Wang and H. Wu, J. Hazard. Mater., 2006, 136, 482–501 CrossRef CAS.
- K. S. Hui and C. Y. H. Chao, Microporous Mesoporous Mater., 2006, 88, 145–151 CrossRef CAS.
- H. Tanaka, H. Eguchi, S. Fujimoto and R. Hino, Fuel, 2006, 85, 1329–1334 CrossRef CAS.
- M. Gross-Lorgouilloux, P. Caullet, M. Soulard, J. Patarin, E. Moleiro and I. Saude, Microporous Mesoporous Mater., 2010, 131, 407–417 CrossRef CAS.
- Sindhunata, J. S. J. van Deventer, G. C. Lukey and H. Xu, Ind. Eng. Chem. Res., 2006, 45, 3559–3568 CrossRef CAS.
- J. G. S. van Jaarsveld, J. S. J. van Deventer and G. C. Lukey, Mater. Lett., 2003, 57, 1272–1280 CrossRef CAS.
- S. Wang, L. Li and Z. H. Zhu, J. Hazard. Mater., 2007, 139, 254–259 CrossRef CAS.
- E. Furlani, S. Bruckner, D. Minichelli and S. Maschio, Ceram. Int., 2008, 34, 2137–2142 CrossRef CAS.
- G. Qian, Y. Song, C. Zhang, Y. Xia, H. Zhang and P. Chui, Waste Manage., 2006, 26, 1462–1467 CrossRef CAS.
- S. B. Wang, G. Q. Lu and H. Y. Zhu, Chem. Lett., 1999, 385–386 CrossRef.
- S. Wang and G. Q. Lu, Stud. Surf. Sci. Catal., 2007, 139, 275–280 CrossRef.
- S. B. Wang, Environ. Sci. Technol., 2008, 42, 7055–7063 CrossRef CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- S. Wang, Dyes Pigm., 2008, 76, 714–720 CrossRef CAS.
- M. Pera-Titus, V. Garcia-Molina, M. A. Banos, J. Gimenez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS.
- G. Zhou, H. Sun, S. Wang, H. Ming Ang and M. O. Tadé, Sep. Purif. Technol., 2011, 80, 626–634 CrossRef CAS.
- Q. J. Yang, H. Choi and D. D. Dionysiou, Appl. Catal., B, 2007, 74, 170–178 CrossRef CAS.
- P. Shukla, S. B. Wang, K. Singh, H. M. Ang and M. O. Tade, Appl. Catal., B, 2010, 99, 163–169 CrossRef CAS.
- P. R. Shukla, S. B. Wang, H. Q. Sun, H. M. Ang and M. Tade, Appl. Catal., B, 2010, 100, 529–534 CrossRef CAS.
- W. Zhang, H. L. Tay, S. S. Lim, Y. S. Wang, Z. Y. Zhong and R. Xu, Appl. Catal., B, 2010, 95, 93–99 CrossRef CAS.
- P. Shukla, H. Sun, S. Wang, H. M. Ang and M. O. Tadé, Catal. Today, 2011, 175, 380–385 CrossRef CAS.
- P. Shukla, H. Q. Sun, S. B. Wang, H. M. Ang and M. O. Tade, Sep. Purif. Technol., 2011, 77, 230–236 CrossRef CAS.
- H. Sun, H. Tian, Y. Hardjono, C. E. Buckley and S. Wang, Catalysis Today, 2011 DOI:10.1016/j.cattod.2011.1009.1001 in press.
- Q. J. Yang, H. Choi, Y. J. Chen and D. D. Dionysiou, Appl. Catal., B, 2008, 77, 300–307 CrossRef CAS.
- T. A. Zepeda, T. Halachev, B. Pawelec, R. Nava, T. Klimova, G. A. Fuentes and J. L. G. Fierro, Catal. Commun., 2006, 7, 33–41 CrossRef CAS.
- R. Xu and H. C. Zeng, Langmuir, 2004, 20, 9780–9790 CrossRef CAS.
- H. Liang, Y. Y. Ting, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, J. Colloid Interface Sci., 2012, 372, 58–62 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |