Ambient temperature synthesis of narrow or monodisperse, highly cross-linked, and “living” polymer microspheres by atom transfer radical precipitation polymerization†
Jingshuai
Jiang
,
Ying
Zhang
,
Xianzhi
Guo
and
Huiqi
Zhang
*
Key Laboratory of Functional Polymer Materials (Nankai University), Ministry of Education, Department of Chemistry, Nankai University, Tianjin 300071, People's Republic of China. E-mail: zhanghuiqi@nankai.edu.cn; Fax: +86-2223507193; Tel: +86-2223507193
First published on 17th May 2012
Abstract
A facile ambient temperature atom transfer radical precipitation polymerization (ATRPP) approach is developed for the efficient one-pot synthesis of narrow or monodisperse, highly cross-linked, and “living” polymer microspheres under mild reaction conditions. The simple introduction of an atom transfer radical polymerization (ATRP) mechanism into a precipitation polymerization system, together with the rational use of polar alcoholic solvents, allows the direct ambient temperature preparation of uniform “living” polymer microspheres with their number-average diameters ranging from 0.36–1.95 μm and their particle size distributions being typically less than 1.01. The polymerization parameters (including the monomer loading, polymerization time, and kind of alcoholic solvent) proved to have a pronounced influence on the yields and morphologies of the polymer microspheres, which makes it very convenient to tailor the particle sizes by tuning the polymerization conditions. The general applicability of ambient temperature ATRPP was demonstrated by its successful application in a range of alcoholic solvents as well as its versatility in the synthesis of a series of uniform copolymer microspheres of different monovinyl functional monomers (4-vinylpyridine, glycidyl methacrylate, methyl methacrylate, and 2-hydroxyethyl methacrylate) with ethylene glycol dimethacrylate. In addition, the “livingness” of the resulting polymer microspheres was confirmed by their direct grafting of hydrophilic polymer brushes via surface-initiated ambient temperature ATRP, leading to advanced functional polymer microspheres with significantly improved surface hydrophilicity.
Introduction
Narrow or monodisperse, highly cross-linked, and micrometer-sized spherical polymer particles have drawn considerable interest from both academic and industrial communities in recent years due to their great potential in a wide range of materials science applications.1–5 Among various approaches presently available for preparing such polymer microspheres, precipitation polymerization has received particular attention because of its easy operation and no need for any surfactant or stabilizer.6–22 So far, a great number of uniform highly cross-linked polymer microspheres based on different monovinyl monomers and divinyl cross-linkers have been prepared by this versatile approach, where functional polymer microspheres are particularly interesting because they are highly promising in various advanced application areas.5Surface modification of polymer microspheres by a “grafting from” approach has proven highly efficient for preparing functional polymer microspheres, which permits growing dense functional polymer brushes from the initiating groups on the particle surfaces by using various surface-initiated polymerizations.5,23–27,29–31 Many controlled “grafting from” methods have been developed up to now, where controlled/“living” radical polymerization techniques (CRPs) (e.g., nitroxide-mediated radical polymerization,32 atom transfer radical polymerization (ATRP),33,34 and reversible addition-fragmentation chain transfer (RAFT) polymerization35) have been most widely utilized because of their good control over the polymer architectures, broad applicability to a wide range of monomers, and mild reaction conditions. In all cases, the presence of CRP-“living” groups on the surfaces of polymer particles is necessary. The spherical polymer particles prepared by traditional precipitation polymerization, however, normally don't have such “living” groups, which makes their further surface modification necessary to introduce the required “living” groups prior to their surface functionalization.23–26 Therefore, the development of facile and efficient approaches for the one-pot preparation of narrow or monodisperse, highly cross-linked, and “living” polymer beads is of great importance as it would allow the more efficient synthesis of advanced functional polymer materials with improved quality and expanded applications. In this context, it is worth mentioning that although some approaches have been developed for the preparation of uncross-linked or lightly cross-linked “living” spherical polymer particles36–45 (including the preparation of monodisperse uncross-linked or lightly cross-linked polymer microspheres by two-stage living radical dispersion polymerizations37,41) and for the synthesis of polydisperse, highly cross-linked, and “living” polymer microspheres27,28 by applying various CRPs in dispersed polymerization systems, the versatile ones for the preparation of narrow or monodisperse, highly cross-linked, and “living” polymer microspheres are still rare.
Very recently, we reported a facile and efficient one-pot approach to obtaining narrow or monodisperse, highly cross-linked, and “living” polymer microspheres by carrying out atom transfer radical precipitation polymerization (ATRPP) in acetonitrile at 60 °C through the introduction of the ATRP mechanism into a precipitation polymerization system, which paves the way for the efficient preparation of advanced functional “living” polymer microspheres with a great potential for various materials science applications.46 Herein, we present our recent progress in the development of ambient temperature ATRPP for the more efficient synthesis of uniform, highly cross-linked, and “living” polymer microspheres (Scheme 1). In comparison with our previously reported ATRPP approach (which needs to be performed at elevated temperatures), the new approach presented here can be easily carried out under very mild ambient temperature conditions by simply using alcoholic solvents in an ATRPP system, which is not only of great importance from environmental and commercial viewpoints,47–50 but also highly necessary for monomers susceptible to elevated temperatures.49,50 Note that although significant progress has been made in the field of precipitation polymerization and some efficient precipitation polymerization approaches have been developed for the synthesis of uniform highly cross-linked polymer microspheres, such as traditional precipitation polymerization7–12,14–16,19,20,22 and distillation precipitation polymerization,13 they are all required to be conducted at elevated temperatures (normally 60–90 °C). In addition, although several ambient temperature or close to ambient temperature precipitation polymerization methods have been described for the synthesis of uniform highly cross-linked polymer microspheres by either γ-ray-induced6,51,52 or photoinduced17,18,21,29 precipitation polymerization, the restricted use of γ-ray sources and UV lamp and the difficulty of γ-ray and UV light in penetrating deeply into the large-scale milky suspension polymerization system considerably limit their broad applications (in particular in large-scale preparation of uniform polymer microspheres). In contrast, our ambient temperature ATRPP can solve the above problems. This, together with its high controllability, permits the more efficient synthesis of narrow or monodisperse, highly cross-linked, and “living” polymer microspheres under mild and easily scalable conditions. To our knowledge, the findings we report here represent the first successful example of the ambient temperature synthesis of uniform highly cross-linked polymer microspheres in the absence of any external polymerization-initiating source (i.e., γ-ray and UV light). The effects of the polymerization parameters (including the monomer loading, polymerization time, and kind of alcoholic solvent) on the morphologies and yields of the polymer particles were studied. In addition, the general applicability of ambient temperature ATRPP and the “livingness” of the obtained polymer microspheres were also demonstrated.
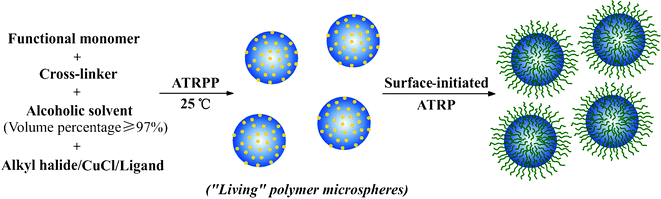 | ||
| Scheme 1 The schematic protocol for the synthesis of narrow or monodisperse, highly cross-linked, and “living” polymer microspheres by ambient temperature ATRPP and their subsequent grafting of hydrophilic polymer brushes via surface-initiated ATRP of functional monomers. | ||
Experimental
Materials
4-Vinylpyridine (4-VP, Alfa Aesar, 96%) and ethylene glycol dimethacrylate (EGDMA, Alfa Aesar, 98%) were purified by distillation under vacuum. Glycidyl methacrylate (GMA, Tianjin heowns Biochemical Technology Co., Ltd., China, 97%) was purified by passing through a neutral aluminium oxide column. Methyl methacrylate (MMA, Sinopharm Chemical Reagent Co., Ltd., Shanghai, China, chemical pure (CP)) was purified by distillation under vacuum. 2-Hydroxyethyl methacrylate (HEMA, Tianjin Institute of Chemical Reagents, China, CP) was purified by washing its aqueous solution (25 vol% of HEMA) with hexane (4 × 200 mL), salting it out of the aqueous phase by addition of NaCl, drying over MgSO4, and distilling under reduced pressure. N-Isopropylacrylamide (NIPAAm, Acros, 99%) was purified by recrystallization from hexane. Copper(I) chloride (CuCl, Tianjin Jiangtian Chemical Technology Co., Ltd., China, analytical reagent (AR)) was purified by stirring it with acetic acid for 12 h, washing with ethanol and diethyl ether, and then dried under vacuum at 75 °C for 3 days. Tris[2-(dimethylamino)ethyl]amine (Me6TREN) was prepared from commercially available tris(2-aminoethyl)amine (Acros, 97%) according to the reported one-step procedure.53 Methanol (Tianjin Jiangtian Chemical Technology Co., Ltd., AR), Ethanol (Tianjin Jiangtian Chemical Technology Co., Ltd., AR), 1-propanol (Tianjin Jiangtian Chemical Technology Co., Ltd., AR), 2-propanol (or isopropanol, Tianjin Jiangtian Chemical Technology Co., Ltd., AR), 1-butanol (Tianjin Jiangtian Chemical Technology Co., Ltd., AR), 1-pentanol (Tianjin Jiangtian Chemical Technology Co., Ltd., AR), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, Aldrich, 99%), 2,2′-bipyridine (Tianjin Jiangtian Chemical Technology Co., Ltd., 99.5+%), anhydrous copper(II) chloride (CuCl2, Alfa Aesar, 98%), copper(II) bromide (CuBr2, Alfa Aesar, 99%), ethyl 2-chloropropionate (Alfa Aesar, 97%), and all the other chemicals were used as received unless otherwise stated.Preparation of highly cross-linked poly(4-VP-co-EGDMA) microspheres by ambient temperature ATRPP
The recipes for the ambient temperature ATRPP of 4-VP with EGDMA in different alcohols are listed in Table 1 (entries 1–35). A typical procedure for the preparation of poly(4-VP-co-EGDMA) microspheres (sample entry 2a) is presented as follows: To a one-neck round-bottom flask (50 mL) with a magnetic stir bar inside, CuCl (1.56 mg, 0.0157 mmol), 4-VP (36.84 mg, 0.3504 mmol), EGDMA (277.56 mg, 1.4015 mmol), and 2-propanol (30 mL) were added successively. The reaction mixture was purged with argon for 15 min and then PMDETA (5.46 mg, 0.0315 mmol) was added. After another 15 min of argon bubbling, ethyl 2-chloropropionate (2.16 mg, 0.0157 mmol) was added into the system. The flask was then sealed, immersed into a thermostated water bath at 25 °C, and stirred for 24 h with a magnetic stir bar at a stirring rate of 90 rpm. The resulting polymer particles were separated by centrifugation and washed with methanol several times to remove the copper catalyst, which were then dried at 40 °C under vacuum overnight to provide a white powder (yield: 17%).| Entryb | Solvent | EGDMA/FM/initiator/CuCl/PMDETAc (molar ratio) | FMc + EGDMA (vol%) | Polym time (h) | yieldd (%) | D n e (μm) | U e | CVe (%) |
|---|---|---|---|---|---|---|---|---|
| a All the polymerizations were performed at 25 °C under magnetic stirring conditions (90 rpm) with the amount of the solvent used being 30 mL except entry 23 (its solvent amount is 120 mL, i.e., the reaction scale of entry 23 is 4 times as large as that of entry 20). b Entries 2a and 2b are the repetition experiments, so are entries 5a, 5b, and 5c. c FM refers to the monovinyl functional monomer, which is 4-VP for entries 1–35, GMA for entry 36, MMA for entry 37, and HEMA for entry 38, respectively, and the initiator is ethyl 2-chloropropionate. d The data without a bracket behind them refer to the yields of the polymer particles obtained from the polymerization solutions and negligible amounts of precipitated polymer particles were observed at the end of the polymerization in such systems, while the data outside and inside the brackets refer to the yields of the polymer particles obtained from the polymerization solutions and those precipitated onto the bottom of the reaction flasks during the polymerization processes, respectively. e D n, U, and CV are the number-average diameter, polydispersity index, and coefficient of variation of the polymer microspheres, respectively; The Dn, U, and CV data listed in the table are those of the polymer microspheres obtained from the polymerization solutions instead of the precipitated particles, except entry 18. | ||||||||
| 1 | 2-Propanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 15 | 0.68 | 1.004 | 3.7 |
| 2a | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 24 | 17 | 0.75 | 1.004 | 3.8 |
| 2b | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 24 | 18 | 0.76 | 1.004 | 3.8 |
| 3 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.2 | 24 | 18 | 0.83 | 1.005 | 4.2 |
| 4 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.4 | 24 | 19 | 0.86 | 1.005 | 4.2 |
| 5a | 2-Propanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 21 | 1.22 | 1.004 | 3.7 |
| 5b | 2-Propanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 20 | 1.21 | 1.004 | 3.7 |
| 5c | 2-Propanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 20 | 1.22 | 1.003 | 3.5 |
| 6 | 2-Propanol | 100/25/1.125/1.125/2.25 | 3.0 | 24 | 17 (9) | 1.95 | 1.005 | 4.0 |
| 7 | 2-Propanol | 100/25/1.125/1.125/2.25 | 3.0 | 7 | 16 | 1.19 | 1.003 | 3.2 |
| 8 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 3 | 2 | 0.36 | 1.017 | 8.5 |
| 9 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 4 | 3 | 0.41 | 1.008 | 5.3 |
| 10 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 6 | 7 | 0.54 | 1.006 | 4.5 |
| 11 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 12 | 12 | 0.66 | 1.004 | 3.7 |
| 12 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 18 | 15 | 0.70 | 1.004 | 3.6 |
| 13 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 24 | 18 | 0.76 | 1.004 | 3.8 |
| 14 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 30 | 21 | 0.81 | 1.004 | 3.8 |
| 15 | 2-Propanol | 100/25/1.125/1.125/2.25 | 1.0 | 36 | 27 | 0.87 | 1.004 | 3.5 |
| 16 | Methanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 57 | 0.76 | 1.005 | 4.0 |
| 17 | Methanol | 100/25/1.125/1.125/2.25 | 1.4 | 24 | 60 | 1.07 | 1.004 | 3.5 |
| 18 | Methanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 0 (60) | 1.32 | 1.059 | 14.2 |
| 19 | Methanol | 100/25/1.125/1.125/2.25 | 2.0 | 7 | 34 | 1.01 | 1.004 | 3.6 |
| 20 | Ethanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 39 | 0.73 | 1.005 | 4.0 |
| 21 | Ethanol | 100/25/1.125/1.125/2.25 | 1.4 | 24 | 41 | 1.06 | 1.005 | 4.2 |
| 22 | Ethanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 45 | 1.60 | 1.005 | 4.0 |
| 23 | Ethanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 37 | 0.71 | 1.004 | 3.9 |
| 24 | 1-Propanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 37 | 0.90 | 1.009 | 5.9 |
| 25 | 1-Propanol | 100/25/1.125/1.125/2.25 | 1.4 | 24 | 39 | 1.21 | 1.011 | 5.9 |
| 26 | 1-Propanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 22 (18) | 1.90 | 1.005 | 4.4 |
| 27 | 1-Propanol | 100/25/1.125/1.125/2.25 | 2.0 | 7 | 22 | 1.62 | 1.005 | 3.9 |
| 28 | 1-Butanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 28 | 0.85 | 1.007 | 4.7 |
| 29 | 1-Butanol | 100/25/1.125/1.125/2.25 | 1.4 | 24 | 30 | 1.11 | 1.004 | 3.6 |
| 30 | 1-Butanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 18 (15) | 1.56 | 1.008 | 5.3 |
| 31 | 1-Butanol | 100/25/1.125/1.125/2.25 | 2.0 | 7 | 16 | 1.08 | 1.006 | 4.5 |
| 32 | 1-Pentanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 23 | 0.85 | 1.004 | 3.8 |
| 33 | 1-Pentanol | 100/25/1.125/1.125/2.25 | 1.4 | 24 | 26 | 1.00 | 1.006 | 4.5 |
| 34 | 1-Pentanol | 100/25/1.125/1.125/2.25 | 2.0 | 24 | 11 (18) | 1.40 | 1.005 | 4.0 |
| 35 | 1-Pentanol | 100/25/1.125/1.125/2.25 | 2.0 | 7 | 13 | 1.06 | 1.007 | 4.7 |
| 36 | Ethanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 41 | 0.96 | 1.003 | 3.4 |
| 37 | Methanol | 100/25/1.125/1.125/2.25 | 0.8 | 24 | 32 | 1.14 | 1.004 | 3.5 |
| 38 | Methanol | 100/25/1.125/1.125/2.25 | 0.6 | 7 | 19 | 1.28 | 1.007 | 4.8 |
A series of other highly cross-linked poly(4-VP-co-EGDMA) microspheres were also prepared similarly following the above procedure by changing the polymerization parameters, including the monomer loading, polymerization time, kind of alcoholic solvent, and reaction scale.
Preparation of highly cross-linked poly(GMA-co-EGDMA), poly(MMA-co-EGDMA), and poly(HEMA-co-EGDMA) microspheres by ambient temperature ATRPP
The recipes for the ambient temperature ATRPP of GMA, MMA, or HEMA with EGDMA are listed in Table 1 (entries 36–38). A typical procedure for the preparation of poly(GMA-co-EGDMA) microspheres is presented as follows (entry 36): To a one-neck round-bottom flask (50 mL) with a magnetic stir bar inside, CuCl (1.30 mg, 0.01313 mmol), GMA (41.49 mg, 0.2919 mmol), EGDMA (231.22 mg, 1.1675 mmol), and ethanol (30 mL) were added successively. The reaction mixture was purged with argon for 15 min and then PMDETA (4.56 mg, 0.02631 mmol) was added. After another 15 min of argon bubbling, ethyl 2-chloropropionate (1.80 mg, 0.01313 mmol) was added into the system. The flask was then sealed, immersed into a thermostated water bath at 25 °C, and stirred for 24 h with a magnetic stir bar at a stirring rate of 90 rpm. The resulting polymer particles were separated by centrifugation and washed with methanol several times to remove the copper catalyst, which were then dried at 40 °C under vacuum overnight (yield: 41%).Highly cross-linked poly(MMA-co-EGDMA) (Table 1, entry 37) and poly(HEMA-co-EGDMA) (Table 1, entry 38) microspheres were also prepared following the above experimental procedure by using the polymerization conditions listed in Table 1.
Grafting polymer brushes onto polymer microspheres by surface-initiated ambient temperature ATRP
Poly(NIPAAm) brushes were grafted onto poly(4-VP-co-EGDMA) microspheres according to the following procedure: polymer microspheres with surface-bound ATRP initiating groups (i.e., the sample entry 23 in Table 1) (50 mg), NIPAAm (0.34 g, 3.00 mmol), CuCl (5.05 mg, 0.0510 mmol), CuCl2 (0.67 mg, 0.0049 mmol), and 2-propanol (2 mL) were successively added into a one-neck round-bottom flask (25 mL). After the reaction mixture was degassed by bubbling argon for 15 min in an ice-bath, Me6TREN (12.46 mg, 0.0544 mmol) was added. After another 15 min of argon bubbling, the reaction mixture was sealed. The polymerization was performed at ambient temperature with stirring for 24 h. After centrifugation, the resulting solid product was thoroughly washed with methanol to remove the copper catalyst and then dried at 25 °C under vacuum to a constant weight, leading to the grafted polymer microspheres with a weight of 54.5 mg.Poly(HEMA) brushes were grafted onto the polymer microspheres according to the following procedure: polymer microspheres with surface-bound ATRP initiating groups (the sample entry 23 in Table 1) (50 mg), HEMA (0.95 g, 7.30 mmol), CuCl (7.21 mg, 0.0728 mmol), CuBr2 (4.86 mg, 0.0217 mmol), methanol (1 mL), and distilled water (1 mL) were successively added into a one-neck round-bottom flask (25 mL). After the reaction mixture was degassed by bubbling argon for 15 min in an ice-bath, 2,2′-bipyridine (31.90 mg, 0.2042 mmol) was added. After another 15 min of argon bubbling, the reaction mixture was sealed. The polymerization was carried out at ambient temperature with stirring for 24 h. After centrifugation, the resulting solid product was thoroughly washed with methanol to remove the copper catalyst and then dried at 25 °C under vacuum to a constant weight, leading to the grafted polymer microspheres with a weight of 55.4 mg.
Characterizations
Fourier Transform Infrared (FT-IR) spectra of the polymer microspheres were measured with a Bio-Rad FTS-6000 spectrometer.The morphologies, particle sizes, and size distributions of the polymer microspheres were determined with a scanning electron microscope (SEM, Shimadzu SS-550). All of the SEM size data reflect the averages of more than 100 particles, which are calculated by the following formulas:7,46
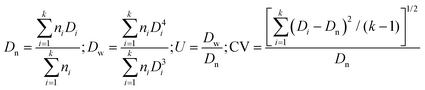
where Dn is the number-average diameter, Dw the weight-average diameter, U the size distribution index (or particle size distribution), CV the coefficient of variation, k the total number of the measured particles, Di the particle diameter of the ith polymer microsphere, and ni the particle number of the microspheres with a diameter Di.
The dispersion properties of the polymer microspheres in pure water were studied as follows: the suspensions of the polymer microspheres in pure water (1 mg mL−1) were first dispersed by ultrasonic, and they were then allowed to settle down for a certain time at 25 °C to check their dispersion stability.
The static contact angle measurements for the polymer films were performed according to the following procedure: the films of the polymer microspheres were prepared by casting their suspension solutions in N,N-dimethylformamide (10 mg mL−1, after ultrasonic dispersion) on clean glass surfaces. After the solvent was allowed to evaporate at ambient temperature overnight, the formed polymer films were further dried under vacuum for 24 h. A KRÜSS FM40 Easy Drop contact angle equipment (Germany) was utilized to determine their static water contact angles. Three measurements were taken across each sample, with their average being used for analysis.
Results and Discussion
ATRPP involves the introduction of the ATRP mechanism into a precipitation polymerization system, which can be easily implemented by simply replacing the initiator normally used in the traditional precipitation polymerization (e.g., AIBN) with an ATRP initiating system (e.g., an alkyl halide and a transition-metal complex formed by a transition metal in its lower oxidation state and a ligand33,34).46 In a recent paper, we have successfully synthesized narrow or monodisperse, highly cross-linked, and “living” polymer microspheres by performing ATRPP in acetonitrile at 60 °C.46 To make ATRPP a more robust approach for the preparation of such advanced functional polymer microspheres under mild reaction conditions, we aim to develop ambient temperature ATRPP in the present work, which is not only of great environmental and commercial importance, but also highly necessary for the monomers sensitive to elevated temperatures. The effects of polymerization parameters (including the monomer loading, polymerization time, and kind of solvent), the general applicability of ambient temperature ATRPP, and the “livingness” of the resulting polymer microspheres were studied in the following parts.Ambient temperature ATRPP in 2-propanol
The solvent used in precipitation polymerization has proven to play a critical role in the successful preparation of narrow or monodisperse highly cross-linked polymer microspheres. An important standard for the choice of an appropriate solvent for a precipitation polymerization system is that it can dissolve all the reactants and in the meantime meets the requirement of being a near θ solvent for the formed polymers. So far, acetonitrile has been the predominantly utilized solvent in precipitation polymerization, which has a solubility parameter (δ) of 24.3 MPa1/2.54 Since acetonitrile has been successfully utilized in our previous ATRPP system for the synthesis of narrow or monodisperse, highly cross-linked, and “living” polymer microspheres at 60 °C,46 a solvent with a similar δ value to acetonitrile, which at the same time can allow the implementation of ambient temperature ATRP, should be the choice of ambient temperature ATRPP. In this respect, 2-propanol appears to meet the above requirements because it has a δ value of 23.5 MPa1/2.54 Most importantly, it has been demonstrated that controlled ambient temperature ATRP can be performed for a range of monomers in 2-propanol55–58 (or in other alcohols59,60). Therefore, 2-propanol was first selected as the polymerization solvent to check whether ambient temperature ATRPP can be realized in such a polar medium. It is worth mentioning here that in comparison with the harmful acetonitrile,19 2-propanol is not only relatively non-toxic (it has been used widely as a solvent for coatings, industrial processes, and pharmaceutical applications and as a cleaning fluid for electronic devices, as described by Wikipedia, the free encyclopedia (http://en.wikipedia.org/wiki/Isopropyl_alcohol)) but also cheaper, which makes it a cost-effective and environmentally-friendly solvent for the studied ATRPP system.A model system was chosen here to demonstrate the proof-of-principle, which utilized 4-VP, EGDMA, ethyl 2-chloropropionate, CuCl, PMDETA, and 2-propanol as the monovinyl functional monomer, divinyl cross-linking monomer, initiator, transition metal salt, ligand, and solvent, respectively. The use of the monovinyl functional monomer in the above polymerization system can lead to copolymer microspheres with extra surface-immobilized functional groups, which should make them more useful for many applications. For example, gold-immobilized polymer microspheres have been prepared by using the polymer particles with pyridine groups.39
An ambient temperature ATRPP experiment was then carried out in 2-propanol at 25 °C for 24 h under magnetic stirring (90 rpm) with a reactant composition of EGDMA/4-VP/ethyl 2-chloropropionate/CuCl/PMDETA being 100/25/1.125/1.125/2.25 (molar ratio) and the total volume of EGDMA and 4-VP being 1.0 vol% of the whole reaction medium (Table 1, entry 2a). SEM characterization revealed that monodisperse poly(4-VP-co-EGDMA) microspheres were formed, which had a number-average diameter Dn of 0.75 μm, a particle size distribution U of 1.004, and a coefficient of variation CV of 3.8% (Fig. 1b). The above results strongly demonstrated that ambient temperature ATRPP could indeed be successfully realized in 2-propanol for the synthesis of uniform highly cross-linked polymer microspheres. Note that a repetition ambient temperature ATRPP experiment was performed for the above polymerization system and totally reproducible data were obtained (entries 2a,b in Table 1, Figs. S1a,b in Electronic Supplementary Information†), thus indicating the reliability of the experimental data. Encouraged by these positive results, we further studied the effects of monomer loading and polymerization time on the ambient temperature ATRPP in 2-propranol.
 | ||
| Fig. 1 SEM images of poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP in 2-propanol for 24 h with a monomer loading of 0.8 (a), 1.0 (b), 1.2 (c), 1.4 (d), 2.0 (e), and 3.0 vol% (f), respectively (the samples a, b, c, d, e, and f correspond to entries 1, 2a, 3, 4, 5a, and 6 in Table 1, respectively). The scale bar corresponds to 1 μm in parts a–f. | ||
To get more insight into the ambient temperature ATRPP, a series of time-dependent ATRPP experiments were carried out in 2-propanol at 25 °C to trace the whole polymerization process (Table 1, entries 8–15). The polymerization solutions were found to turn from a transparent clear solution to a slightly turbid state at a polymerization time of about 2.5 h, indicating the occurrence of the particle nucleation process. Fig. 2 shows the SEM images of poly(4-VP-co-EGDMA) microspheres obtained at different polymerization times. It is interesting to note that polymer microspheres with rather uniform sizes (U ≤ 1.017) were obtained even at the beginning of the polymerization (just as observed in our previous ATRPP system performed at the elevated temperature46), which is considerably different from many traditional precipitation polymerization systems, where highly polydisperse uneven polymer particles were produced at the early stage of the polymerization.18,19,21 In addition, the yields, sizes, and uniformity of the polymer microspheres increased with increasing the polymerization time. For example, the yields of the polymer microspheres increased from 2 to 27% with increasing the polymerization time from 3 to 36 h; the Dn and U values of the polymer microspheres obtained at a polymerization time of 3 h were 0.36 μm and 1.017, respectively, while those of the polymer microspheres obtained at a polymerization time of 36 h were 0.87 μm and 1.004, respectively. More importantly, a very good linear relationship between the yields and the cubes of Dn of the polymer microspheres was obtained for the studied system (Fig. 3), demonstrating that the number of polymer particles remained constant during the ambient temperature ATRPP process and neither coagulation nor secondary nucleation occurred after the nucleation process.13,46
 | ||
| Fig. 2 SEM images of poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP in 2-propanol at a polymerization time of 3 (a), 4 (b), 6 (c), 12 (d), 18 (e), 24 (f), 30 (g), and 36 h (h), respectively (monomer loading = 1.0 vol%) (the samples a–h correspond to entries 8–15 in Table 1, respectively). The scale bar corresponds to 1 μm in parts a–h. | ||
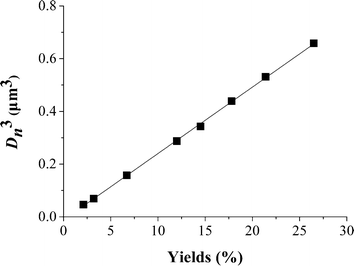 | ||
| Fig. 3 Plot of the cubes of the number-average diameters of poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP in 2-propanol at different polymerization times versus their yields. | ||
Fig. 4a presents the FT-IR spectra of poly(4-VP-co-EGDMA) microspheres obtained at different polymerization times, from which the incorporated levels of EGDMA and 4-VP in the polymer microspheres can be readily derived by comparing the peak height for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band from the bonded EGDMA (1731 cm−1) with that for the CN stretching band (around 1597 cm−1) or CC stretching band (1456 cm−1) from the bonded 4-VP (Fig. 4b).30,46 It can be seen clearly that the amount of the bonded EGDMA relative to the bonded 4-VP in the polymer microspheres decreased with increasing the polymerization time at the early stage of the ambient temperature ATRPP process, while it leveled off at a polymerization time ≥6 h. The above results suggested that a relatively higher level of EGDMA might be incorporated into the polymer particles during the nucleation process (in comparison with that incorporated during the particle growth process), while homogeneous grafting of a constant amount of EGDMA and 4-VP molecules onto the polymer microspheres took place during most of the particle growth period, which should lead to the formation of a uniformly cross-linked network around the polymer nuclei in the polymer microspheres prepared by ambient temperature ATRPP,36,61–63 just as observed in our previously reported ATRPP system.46 Further investigation is ongoing to experimentally confirm the homogeneity of the cross-linked networks of these polymer microspheres. Note that the uniform grafting of EGDMA and 4-VP molecules onto the polymer microspheres during the particle growth process in ambient temperature ATRPP might provide polymer microspheres with a more compact surface layer in comparison with those prepared via traditional precipitation polymerization (which have a lightly cross-linked transient solvent-swollen gel layers on the particle surfaces due to their capturing of soluble oligomers8). Therefore, the polymer particles generated in ambient temperature ATRPP should be less stabilized in the reaction media than those generated in traditional precipitation polymerization, which might account for the relatively lower monomer loadings in ambient temperature ATRPP for the preparation of narrow or monodisperse polymer microspheres.
O band from the bonded EGDMA (1731 cm−1) with that for the CN stretching band (around 1597 cm−1) or CC stretching band (1456 cm−1) from the bonded 4-VP (Fig. 4b).30,46 It can be seen clearly that the amount of the bonded EGDMA relative to the bonded 4-VP in the polymer microspheres decreased with increasing the polymerization time at the early stage of the ambient temperature ATRPP process, while it leveled off at a polymerization time ≥6 h. The above results suggested that a relatively higher level of EGDMA might be incorporated into the polymer particles during the nucleation process (in comparison with that incorporated during the particle growth process), while homogeneous grafting of a constant amount of EGDMA and 4-VP molecules onto the polymer microspheres took place during most of the particle growth period, which should lead to the formation of a uniformly cross-linked network around the polymer nuclei in the polymer microspheres prepared by ambient temperature ATRPP,36,61–63 just as observed in our previously reported ATRPP system.46 Further investigation is ongoing to experimentally confirm the homogeneity of the cross-linked networks of these polymer microspheres. Note that the uniform grafting of EGDMA and 4-VP molecules onto the polymer microspheres during the particle growth process in ambient temperature ATRPP might provide polymer microspheres with a more compact surface layer in comparison with those prepared via traditional precipitation polymerization (which have a lightly cross-linked transient solvent-swollen gel layers on the particle surfaces due to their capturing of soluble oligomers8). Therefore, the polymer particles generated in ambient temperature ATRPP should be less stabilized in the reaction media than those generated in traditional precipitation polymerization, which might account for the relatively lower monomer loadings in ambient temperature ATRPP for the preparation of narrow or monodisperse polymer microspheres.
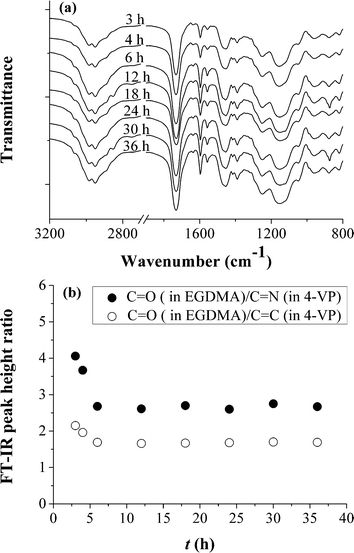 | ||
| Fig. 4 (a) FT-IR spectra of poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP in 2-propanol at different polymerization times; (b) FT-IR peak height ratio of CO stretching band from the bonded EGDMA to CN or CC stretching band from the bonded 4-VP (which refers to the bonded amount of EGDMA relative to that of 4-VP) in poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP at different polymerization times. | ||
Ambient temperature ATRPP in other alcoholic solvents
To evaluate the scope of ambient temperature ATRPP and demonstrate its general applicability, a series of other alcohols (including methanol, ethanol, 1-propanol, 1-butanol, and 1-pentanol) were also utilized as the polymerization solvents for the ambient temperature ATRPP of 4-VP with EGDMA and their effects on the polymerization processes were studied. The ambient temperature ATRPP experiments were conducted at 25 °C in different alcohols with the polymerization parameters, such as the reactant composition, volume of the solvent, and stirring rate being held constant, where the monomer loadings were varied from 0.8 to 2.0 vol% in each solvent system (Note that all the above ambient temperature ATRPP experiments were performed for 24 h. In addition, a shorter polymerization time of 7 h was also applied for those ATRPP systems with the occurrence of obvious precipitation of polymer particles onto the bottom of the reaction flasks after 24 h of polymerization) (Table 1, entries 16–22,24–35). The experimental results showed that uniform polymer microspheres could be readily obtained under most of the studied polymerization conditions (Fig. 5, Fig. S4†) although the utilized solvents showed a pronounced influence on the ambient temperature ATRPP processes (Table 1). The polymerization rates proved to decrease with increasing the alkyl chain length in the alcohols from methyl to pentyl groups, as indicated by the yields of the resulting polymer particles at the same level of monomer loadings and the same polymerization times. This could be attributed to the reduced polarity of alcohols with increasing their alkyl chain length because the solvents with higher polarity have proven to be capable of accelerating the polymerization rates of the ATRP systems.64–67 In addition, both the sizes of the polymer microspheres obtained from the polymerization solutions and the total yields of polymer particles (including the polymer particles obtained from the polymerization solutions and those precipitated onto the bottom of the reaction flasks in case of the occurrence of obvious precipitation of polymer particles during the ATRPP processes) increased with increasing the monomer loadings in each solvent system under the similar reaction conditions, just as observed in the above-described ambient temperature ATRPP system with 2-propanol as the solvent and in our previously reported ATRPP system.46 It is noteworthy that significant amounts of precipitated polymer particles were observed on the bottom of the reaction flasks at a polymerization time of 24 h when the monomer loadings were 2.0 vol% for the ambient temperature ATRPP system with methanol, 1-propanol, 1-butanol, or 1-pentanol as the solvent (Figs. S4a3 and S2d,f,h†), while the amounts of the precipitated polymer particles were negligible for the ambient temperature ATRPP system with ethanol or 2-propanol as the solvent under the otherwise same reaction conditions. The real cause is not very clear yet at this stage, but might be attributed to the cooperative effects of several influencing factors such as the polarity and viscosity of the solvents. Shortening the polymerization time to 7 h in the above reactions, however, resulted in polymer microspheres with low dispersity with negligible precipitated polymer particles (Figs. S3b–e†, entries 19, 27, 31, and 35 in Table 1). It is also worth noting here that the ambient temperature ATRPP with 1-propanol as the solvent showed considerably higher polymerization rates than those performed in 2-propanol under the otherwise same polymerization conditions, which might be ascribed to the relatively higher polarity of 1-propanol (the polar Hansen parameter δP = 6.8 MPa1/2)54 in comparison with 2-propanol (δP = 6.1 MPa1/2).54 | ||
| Fig. 5 SEM images of poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP for 24 h in different alcohols including methanol (a), ethanol (b), 1-propanol (c), 1-butanol (d), and 1-pentanol (e), respectively (monomer loading = 1.4 vol%) (the samples a, b, c, d, and e correspond to entries 17, 21, 25, 29, and 33 in Table 1, respectively). The scale bar corresponds to 1 μm in parts a–e. | ||
In this context, it should be interesting to compare the ambient temperature ATRPP with our previously reported ATRPP (which were carried out at 60 °C).46 The experimental results showed that relatively high yields of polymer microspheres were obtained for the ambient temperature ATRPP in such alcoholic solvents as methanol, ethanol, and 1-propanol, which are comparable with those of our previous ATRPP system46 (but they are much higher than those of the photoinduced precipitation polymerizations performed at ambient temperature17,18,21). In addition, the polymer microspheres prepared by ambient temperature ATRPP proved to have smaller particle sizes than those prepared by ATRPP at 60 °C,46 which might be due to the decrease in the solubility of the initially formed branched oligomers with decreasing the polymerization temperature (i.e., the oligomers with relatively lower molecular weights became insoluble in the ambient temperature reaction media), leading to an increase in the amount of particle nuclei and thus smaller particle sizes. In addition, the decreased polymerization rates at lower temperatures might also be partially responsible for the above phenomenon. This explanation has been experimentally verified by performing ATRPP in an alcoholic solvent (i.e., 2-propanol) at elevated temperatures, where the particle sizes of the resulting monodisperse polymer microspheres increased with an increase in the polymerization temperature (Fig. S5†).
Ambient temperature ATRPP of other monovinyl functional monomers with EGDMA
Narrow or monodisperse highly cross-linked polymer microspheres with designed surface wettability are of considerable interest because they are highly useful for various materials science applications.5 Such polymer microspheres have been successfully prepared by traditional precipitation polymerization through the introduction of different monovinyl functional monomers into the polymerization system.10,12,15,21 It is thus important to check whether ambient temperature ATRPP is capable of generating uniform copolymer microspheres with different incorporated monovinyl functional monomers. To fulfill this purpose, the ambient temperature ATRPP of several different monovinyl functional monomers (i.e., GMA, MMA, and HEMA) with EGDMA were performed to prepare highly cross-linked copolymer microspheres (Table 1, entries 36–38). Both monodisperse poly(GMA-co-EGDMA) (Fig. 6a, entry 36 in Table 1) and poly(MMA-co-EGDMA) (Fig. 6b, entry 37 in Table 1) microspheres were readily obtained following a similar procedure as that used for the synthesis of poly(4-VP-co-EGDMA) microspheres, whereas uniform poly(HEMA-co-EGDMA) microspheres (Fig. 6c, entry 38 in Table 1) were only formed at a lower monomer loading (i.e., 0.6%) and a shorter polymerization time (i.e., 7 h). The above results strongly demonstrate that ambient temperature ATRPP is indeed a facile, general, and highly efficient approach to preparing various surface-functionalized copolymer microspheres with uniform sizes. | ||
| Fig. 6 SEM images of poly(GMA-co-EGDMA) (a), poly(MMA-co-EGDMA) (b), poly(HEMA-co-EGDMA) (c), poly(4-VP-co-EGDMA) (d), PHEMA brushes-grafted poly(4-VP-co-EGDMA) (e), and PNIPAAm brushes-grafted poly(4-VP-co-EGDMA) (f) microspheres, respectively (the samples a, b, c, and d correspond to entries 36, 37, 38, and 23 in Table 1, respectively). The scale bar corresponds to 1 μm in parts a–f. | ||
FT-IR was first utilized to confirm the successful incorporation of functional monomers into the above-obtained polymer microspheres. Fig. 7 shows that in addition to the peaks corresponding to the incorporated poly(EGDMA) (i.e., 1731 (CO stretching), 1255 and 1152 cm−1 (C–O–C stretching)), some characteristic peaks of poly(GMA) (around 1233 cm−1, C–O stretching band from epoxy ring) and poly(HEMA) (around 3540 cm−1, O–H stretching band) were also clearly observed in the spectra of poly(GMA-co-EGDMA) and poly(HEMA-co-EGDMA) microspheres, respectively, revealing the presence of the bonded functional monomers in the obtained polymer microspheres. Note that the characteristic absorption peaks of poly(MMA) in poly(MMA-co-EGDMA) microspheres were not discernible due to their overlap with those of poly(EGDMA) in the polymer microspheres.
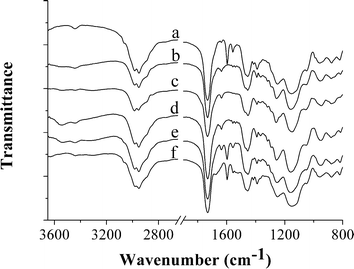 | ||
| Fig. 7 FT-IR spectra of poly(4-VP-co-EGDMA) (a), poly(MMA-co-EGDMA) (b), poly(GMA-co-EGDMA) (c), poly(HEMA-co-EGDMA) (d), PHEMA brushes-grafted poly(4-VP-co-EGDMA) (e), and PNIPAAm brushes-grafted poly(4-VP-co-EGDMA) (f) microspheres, respectively. | ||
It has been well established that the surface wettability of the polymer microspheres can be easily tuned by their incorporation of different functional monomers and it can be accurately evaluated by performing water contact angle experiments.46 Therefore, the static water contact angles of the films prepared with polymer microspheres bearing incorporated 4-VP, GMA, MMA, and HEMA were determined in order to get more insight into their surface properties. Fig. 8a shows the profiles of a water drop on the films of the studied polymer microspheres, from which the static water contact angles of the films prepared with poly(4-VP-co-EGDMA), poly(GMA-co-EGDMA), poly(MMA-co-EGDMA), and poly(HEMA-co-EGDMA) microspheres were determined to be 123, 114, 110, and 109°, respectively. The relatively lower static water contact angles for the films made from poly(GMA-co-EGDMA) poly(MMA-co-EGDMA), and poly(HEMA-co-EGDMA) microspheres in comparison with that prepared with poly(4-VP-co-EGDMA) microspheres further confirmed the successful incorporation of functional monomers into these polymer microspheres.
 | ||
| Fig. 8 (a) Profiles of a water drop on the films prepared with a series of polymer microspheres; (b) Photograph for the dispersion of polymer microspheres in pure water (1 mg mL−1) at 25 °C (after settling down for 3 h). The samples located from left to right in the above two figures are poly(4-VP-co-EGDMA), poly(GMA-co-EGDMA), poly(MMA-co-EGDMA), poly(HEMA-co-EGDMA), PHEMA brushes-grafted poly(4-VP-co-EGDMA), and PNIPAAm brushes-grafted poly(4-VP-co-EGDMA) microspheres, respectively. | ||
The dispersion stability of poly(4-VP-co-EGDMA), poly(GMA-co-EGDMA), poly(MMA-co-EGDMA), and poly(HEMA-co-EGDMA) microspheres in water was also studied to test their surface wettability (Fig. 8b, Fig. S6†). Much faster sedimentation was observed for poly(4-VP-co-EGDMA) microspheres in comparison with poly(GMA-co-EGDMA), poly(MMA-co-EGDMA), and poly(HEMA-co-EGDMA) microspheres, revealing the improved surface hydrophilicity for poly(GMA-co-EGDMA), poly(MMA-co-EGDMA), and poly(HEMA-co-EGDMA) microspheres and thus the successful incorporation of functional monomers into these polymer microspheres.
“Livingness” of the polymer microspheres prepared by ambient temperature ATRPP
It is well known that one of the main advantages that CRPs can offer is their versatility in preparing well-defined polymers with end-capped “living” groups, which makes their further chain extension possible.32–35 Therefore, the ambient temperature ATRPP is expected to provide “living” polymer microspheres with reactive ATRP initiating groups on their surfaces. To confirm this hypothesis, the as-prepared polymer microspheres were utilized as the immobilized ATRP initiator for the surface-initiated ATRP of hydrophilic functional monomers (i.e., NIPAAm and HEMA).The ambient temperature ATRPP of 4-VP with EGDMA with its reaction scale being 4 times as large as that of the entry 20 in Table 1 was first carried out to provide enough polymer microspheres in one batch for the subsequent surface-initiated ATRP (Table 1, entry 23). Note that a round-bottom flask with a volume of 250 mL and an oval-shaped stir bar with its length being 2.8 cm were utilized for this larger scale polymerization in order to have enough room to hold larger volume of polymerization mixture and homogeneously disperse the polymer particles in the reaction medium (while the volume of the round-bottom flask and the length of the oval-shaped stir bar were 50 mL and 1.5 cm for the reaction entry 20, respectively). The experimental results showed that monodisperse polymer microspheres were readily formed in this larger scale polymerization (Fig. 6d) and their particle size distribution U and coefficient of variation CV were almost the same with those of the polymer microspheres obtained in entry 20. However, both the yields and sizes of the polymer microspheres somewhat decreased with an increase in the reaction scale. This might be attributed to the different sizes of the reaction flasks and the stir bars used, which might influence the agitation situation for different reactions, thus leading to their different yields and particle sizes, just as observed in our previous report.46
The surface-initiated ATRP of NIPAAm was then performed in 2-propanol at 25 °C for 24 h under the magnetic stirring with the above-obtained polymer microspheres (sample entry 23 in Table 1: Dn = 0.71 μm, U = 1.004, CV = 3.9%) as the immobilized ATRP initiator and CuCl/CuCl2/Me6TREN as the catalyst. No free initiator was added into the polymerization system, which means that the polymerization should be surface-confined. CuCl2 (CuCl2/CuCl = 0.10, molar ratio) was added into the polymerization solution to improve the controllability of the polymerization system.68,69 A weight increase of 9% was observed for the polymer particles after their surface modification, indicating that PNIPAAm brushes were grafted onto the polymer particles. The SEM investigation showed that the modified polymer particles were still separate microspheres, with their Dn and U values being 0.728 μm and 1.005, respectively (Fig. 6f). An increase of 18 nm in Dn value was observed for the grafted polymer microspheres in comparison with the ungrafted ones, again revealing the successful grafting of PNIPAAm brushes onto the polymer microspheres via the surface-initiated ATRP.
The surface-initiated ATRP of HEMA was carried out in methanol/water (1/1 v/v) at ambient temperature for 24 h under the magnetic stirring with the same sample entry 23 (Table 1) as the immobilized ATRP initiator and CuCl/CuBr2/2,2′-bipyridine as the catalyst. A weight increase of 11% was observed for the polymer particles after their surface modification. The grafted polymer particles also proved to be separate microspheres, with their Dn and U being 0.733 μm and 1.006, respectively (Fig. 6e). An increase of 23 nm in Dn value was observed for the grafted polymer particles in comparison with the ungrafted ones. The above results demonstrate the successful grafting of PHEMA brushes onto the polymer microspheres. It is important to stress here that the increased weights of the grafted polymer microspheres most likely result from the surface-grafted polymer brushes because the use of poor solvents for the “living” polymer microspheres in the grafting polymerization processes and their rather high cross-linking densities would prevent them from swelling in the reaction media and only allow the occurrence of surface polymerization, just as reported by Tirelli and coworkers70 and by our group.46
FT-IR was employed to further verify the successful grafting of PNIPAAm and PHEMA brushes onto the polymer microspheres (Fig. 7). It can be seen clearly that in addition to the absorption peaks from the ungrafted poly(4-VP-co-EGDMA) microspheres, such as those characteristic of the bonded EGDMA (i.e., 1731 (CO stretching) and 1255/1152 cm−1 (C–O–C stretching)) and those corresponding to the CN stretching (1597 and 1557 cm−1) and CC stretching (1456 cm−1) for the bonded 4-VP, some new ones characteristic of PNIPAAm (i.e., the amide I band (1677 cm−1, CO stretching) and amide II band (1529 cm−1, N–H stretching)) and PHEMA (3540 cm−1, O–H stretching) were also observed in the FT-IR spectra of the polymer microspheres obtained via the surface-initiated ATRP of NIPPAm and HEMA, respectively, thus demonstrating the successful grafting of PNIPAAm and PHEMA brushes.
Surface-grafting of hydrophilic polymer brushes has proven to be a highly effective approach to improving the surface hydrophilicity and water dispersion stability of the polymer microspheres.46,71 Therefore, it is envisioned that the polymer particles grafted with PNIPAAm and PHEMA brushes should show much reduced static water contact angles and enhanced dispersion stability in water. The results shown in Fig. 8 indeed support this hypothesis. The static water contact angles of the films prepared with polymer microspheres bearing PNIPAAm and PHEMA brushes were determined to be 65 and 72°, respectively, which were significantly lower than that (123°) of the film prepared with the ungrafted poly(4-VP-co-EGDMA) microspheres. In addition, there was much faster sedimentation for the ungrafted polymer microspheres in water in comparison with the grafted ones (Fig. S6†). These results provide strong evidence for the presence of PNIPAAm and PHEMA brushes on the surfaces of the grafted polymer microspheres.
Conclusions
We have developed a facile, general, and highly efficient ambient temperature ATRPP approach to obtaining narrow or monodisperse, highly cross-linked, surface-functionalized, and “living” polymer microspheres under mild polymerization conditions. Uniform polymer microspheres with their sizes ranging from 0.36–1.95 μm and their particle size distributions being typically lower than 1.01 were readily prepared by simply using alcoholic solvents in ATRPP and their sizes could be easily tailored by tuning the polymerization parameters. The ambient temperature ATRPP proved to be generally applicable in a range of alcoholic solvents and for a number of monovinyl functional monomers, leading to uniform copolymer microspheres with different surface wettability. The “livingness” of the resulting polymer microspheres was also confirmed by their easy surface modification via the surface-initiated ambient temperature ATRP of hydrophilic functional monomers, resulting in advanced functional polymer microspheres with significantly improved surface hydrophilicity. In view of the great environmental and commercial importance of the ambient temperature polymerization techniques and their applicability for the heat-sensitive monomers, the relatively non-toxicity and low prices of alcoholic solvents (e.g., ethanol and 2-propanol), and the high versatility of surface-initiated ATRP (such as its compatibility to a wide range of functional monomers and its very mild reaction conditions) in the controlled modification of various substrate materials, we believe that the ambient temperature ATRPP technique described here represents a general and promising, cost-effective, and environment-friendly methodology for the efficient synthesis of various advanced functional polymer microspheres with a diverse range of application potential in the field of materials science.Acknowledgements
The authors thank the financial support from National Natural Science Foundation of China (20744003, 20774044, 201174067), Natural Science Foundation of Tianjin (11JCYBJC01500), a supporting program for New Century Excellent Talents (Ministry of Education) (NCET-07-0462), and a start-up fund from Nankai University. Professor Yakai Feng (School of Chemical Engineering and Technology, Tianjin University, People's Republic of China) is thanked for his kind help in measuring the static water contact angles of the samples.References
- D. Horák, Acta Polym., 1996, 47, 20–28 CrossRef.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed., 2003, 42, 5336–5338 CrossRef CAS.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- L. Barner, Adv. Mater., 2009, 21, 2547–2553 CrossRef CAS.
- Y. Naka, I. Kaetsu, Y. Yamamoto and K. Hayashi, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1197–1202 CrossRef CAS.
- K. Li and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3257–3263 CrossRef CAS.
- J. Downey, R. S. Frank, W. H. Li and H. D. H. Stöver, Macromolecules, 1999, 32, 2838–2844 CrossRef CAS.
- W. H. Li and H. D. H. Stöver, Macromolecules, 2000, 33, 4354–4360 CrossRef CAS.
- L. Ye, R. Weiss and K. Mosbach, Macromolecules, 2000, 33, 8239–8245 CrossRef CAS.
- J. Downey, G. McIsaac, R. S. Frank and H. D. H. Stöver, Macromolecules, 2001, 34, 4534–4541 CrossRef CAS.
- R. S. Frank, J. Downey, K. Yu and H. D. H. Stöver, Macromolecules, 2002, 35, 2728–2735 CrossRef CAS.
- F. Bai, X. Yang and W. Huang, Macromolecules, 2004, 37, 9746–9752 CrossRef CAS.
- S. E. Shim, S. Yang and S. Choe, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3967–3974 CrossRef CAS.
- J. M. Jin, S. Yang, S. E. Shim and S. Choe, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5343–5346 CrossRef CAS.
- R. Takekoh, W. H. Li, N. A. D. Burke and H. D. H. Stöver, J. Am. Chem. Soc., 2006, 128, 240–244 CrossRef CAS.
- R. Joso, E. H. Pan, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and L. Barner, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3482–3487 CrossRef CAS.
- F. Limé and K. Irgum, Macromolecules, 2007, 40, 1962–1968 CrossRef.
- Q. Yan, Y. Bai, Z. Meng and W. Yang, J. Phys. Chem. B, 2008, 112, 6914–6922 CrossRef CAS.
- Q. Yan, T. Zhao, Y. Bai, F. Zhang and W. Yang, J. Phys. Chem. B, 2009, 113, 3008–3014 CrossRef CAS.
- F. Limé and K. Irgum, Macromolecules, 2009, 42, 4436–4442 CrossRef.
- A. L. Medina-Castillo, J. F. Fernandez-Sanchez, A. Segura-Carretero and A. Fernandez-Gutierrez, Macromolecules, 2010, 43, 5804–5813 CrossRef CAS.
- G. Zheng and H. D. H. Stöver, Macromolecules, 2002, 35, 6828–6834 CrossRef CAS.
- G. Zheng and H. D. H. Stöver, Macromolecules, 2002, 35, 7612–7619 CrossRef CAS.
- G. Zheng and H. D. H. Stöver, Macromolecules, 2003, 36, 1808–1814 CrossRef CAS.
- G. Zheng and H. D. H. Stöver, Macromolecules, 2003, 36, 7439–7445 CrossRef CAS.
- L. Barner, C. Li, X. Hao, M. H. Stenzel, C. Barner-Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5067–5076 CrossRef CAS.
- G. Pan, B. Zu, X. Guo, Y. Zhang, C. Li and H. Zhang, Polymer, 2009, 50, 2819–2825 CrossRef CAS.
- J. Li, B. Zu, Y. Zhang, X. Guo and H. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3217–3228 CrossRef CAS.
- G. Pan, Y. Zhang, X. Guo, C. Li and H. Zhang, Biosens. Bioelectron., 2010, 26, 976–982 CrossRef CAS.
- G. Pan, Y. Ma, Y. Zhang, X. Guo, C. Li and H. Zhang, Soft Matter, 2011, 7, 8428–8439 RSC.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578–5584 CrossRef CAS.
- J. S. Song and M. A. Winnik, Macromolecules, 2006, 39, 8318–8325 CrossRef CAS.
- G. Zheng and C. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS.
- W. Wan and C. Pan, Macromolecules, 2007, 40, 8897–8905 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C. K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS.
- K. Min and K. Matyjaszewski, Macromolecules, 2007, 40, 7217–7222 CrossRef CAS.
- K. J. Thurecht, A. M. Gregory, W. Wang and S. M. Howdle, Macromolecules, 2007, 40, 2965–2967 CrossRef CAS.
- B. Grignard, C. Jérôme, C. Calberg, R. Jérôme, W. Wang, S. M. Howdle and C. Detrembleur, Macromolecules, 2008, 41, 8575–8583 CrossRef CAS.
- K. Min, H. Gao, J. A. Yoon, W. Wu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2009, 42, 1597–1603 CrossRef CAS.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- J. Jiang, Y. Zhang, X. Guo and H. Zhang, Macromolecules, 2011, 44, 5893–5904 CrossRef CAS.
- A. J. Convertine, N. Ayres, C. W. Scales, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2004, 5, 1177–1180 CrossRef CAS.
- L. Lu, N. Yang and Y. Cai, Chem. Commun., 2005, 5287–5288 RSC.
- H. Zheng, W. Bai, K. Hu, R. Bai and C. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2575–2580 CrossRef CAS.
- Y. Kwak and K. Matyjaszewski, Macromolecules, 2010, 43, 5180–5183 CrossRef CAS.
- M. Yoshida, M. Asano, I. Kaetsu and Y. Morita, Radiat. Phys. Chem., 1987, 30, 39–45 CrossRef CAS.
- M. Kumakura, Eur. Polym. J., 1995, 31, 1095–1098 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
- E. A. Grulke, Solubility Parameter Values. in Polymer Handbook, ed. J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, John Wiley & Sons, New York, 4th edn, 1999, pp. VII/675–714 Search PubMed.
- J. Xia, X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 3531–3533 CrossRef CAS.
- X. Chen and S. P. Armes, Adv. Mater., 2003, 15, 1558–1562 CrossRef CAS.
- Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- R. Longenecker, T. Mu, M. Hanna, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2011, 44, 8962–8971 CrossRef CAS.
- K. L. Robinson, M. A. Khan, M. V. de Paz Báñez, X. S. Wang and S. P. Armes, Macromolecules, 2001, 34, 3155–3158 CrossRef CAS.
- X. Bories-Azeau and S. P. Armes, Macromolecules, 2002, 35, 10241–10243 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- W. Huang, G. L. Baker and M. L. Bruening, Angew. Chem., Int. Ed., 2001, 40, 1510–1512 CrossRef CAS.
- B. Zu, G. Pan, X. Guo, Y. Zhang and H. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3257–3270 CrossRef CAS.
- X.-S. Wang, S. F. Lascelles, R. A. Jackson and S. P. Armes, Chem. Commun., 1999, 1817–1818 Search PubMed.
- G. Chambard, B. Klumperman and A. L. German, Macromolecules, 2000, 33, 4417–4421 CrossRef CAS.
- K. Matyjaszewski, H.-J. Paik, P. Zhou and S. J. Diamanti, Macromolecules, 2001, 34, 5125–5131 CrossRef CAS.
- H. Zhang and R. Van der Linde, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3549–3561 CrossRef CAS.
- H. Zhang, B. Klumperman, W. Ming, H. Fischer and R. Van der Linde, Macromolecules, 2001, 34, 6169–6173 CrossRef CAS.
- W. Huang, J. B. Kim, M. L. Bruening and G. L. Baker, Macromolecules, 2002, 35, 1175–1179 CrossRef CAS.
- D. Bontempo, N. Tirelli, K. Feldman, G. Masci, V. Crescenzi and J. A. Hubbell, Adv. Mater., 2002, 14, 1239–1241 CrossRef CAS.
- G. Pan, Y. Zhang, Y. Ma, C. Li and H. Zhang, Angew. Chem., Int. Ed., 2011, 59, 11731–11734 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of poly(4-VP-co-EGDMA) microspheres prepared by ambient temperature ATRPP or ATRPP at elevated temperatures under different polymerization conditions, as well as the detailed photographs for the water dispersion stability of polymer microspheres. See DOI: 10.1039/c2ra01249a |
| This journal is © The Royal Society of Chemistry 2012 |