Study of the spontaneous attachment of polycyclic aryldiazonium salts onto amorphous carbon substrates†
Deirdre M.
Murphy
,
Ronan J.
Cullen
,
Dilushan R.
Jayasundara
,
Eoin M.
Scanlan
and
Paula E.
Colavita
*
School of Chemistry, University of Dublin Trinity College, College Green, Dublin 2, Ireland. E-mail: colavitp@tcd.ie
First published on 11th May 2012
Abstract
Diazonium salts of two nitro-substituted polycyclic aromatic compounds were synthesized and their spontaneous covalent attachment onto amorphous carbon surfaces was studied via electrochemical and spectroscopic techniques. In situ spectroscopic monitoring of the grafting of these compounds at amorphous carbon surfaces via attenuated total internal reflection Fourier transform infrared spectroscopy (ATR-FTIR) highlighted a marked difference in adsorption rates, which was also evident via ex situ electrochemical analysis. We show that adsorption rate differences cannot be explained based on differences in the solvolysis rates of these two molecules. It was found instead that the relative position of the –N2+ groups with respect to the –NO2 groups affected the reduction potential of the diazonium cations and in turn their adsorption rate at amorphous carbon surfaces. We conclude that differences in the electron density at the carbon atom bound to the diazonium group are responsible for the differences observed in the spontaneous attachment at carbon.
Introduction
Aryldiazonium salts have served historically as reagents in transformations such as the Sandmeyer and Heck–Matsuda reactions,1,2 however, in recent years there has been increased interest in their applications for surface modification. Aryldiazonium salts are versatile reagents that can be utilized to modulate the surface chemistry of metals, semiconductors, carbon materials and polymers,3,4 by forming strong covalent linkages between the grafted compound and the substrate. The grafting process can be achieved via electroreduction of the diazonium moiety and the loss of dinitrogen, followed by attachment of the aryl moiety to the surface through a covalent bond.3,5–7 More recently, it was found that covalent attachment could occur spontaneously from solution;8–12 this type of approach is advantageous since it does not require electrical contact to be made to the substrate material and can therefore be applied to a wide range of substrates, including poor conductors, nanostructured materials and nanoparticles in suspension.13–17The spontaneous reaction of aryldiazonium salts on surfaces is mechanistically less well understood than electrografting. The specific substituents on the aryldiazonium, the type of substrate and the solvent used for the deposition have all been found to affect reaction yields and rates.12,18 General trends have been identified showing that electron withdrawing substituents and easily oxidizable metals tend to display faster grafting reactions. Based on these results a mechanism involving the spontaneous electron transfer from metal substrates to aryldiazonium with the loss of dinitrogen has been proposed, in analogy with the electrografting process. The grafting yield on carbon appears to depend, as for metals, on the relative position of the donor and the acceptor levels at the carbon and aryldiazonium cation, respectively, thus suggesting that the surface acts as a mild reductant.19 However, it has been hypothesized that multiple routes could lead to the spontaneous formation of aryldiazonium layers and multilayers.20–23 In particular, it has been proposed that the thermal decomposition of aryldiazonium salts via either homolytic or heterolytic cleavage of C–N2+ bonds gives rise to alternative pathways of the covalent aryldiazonium attachment at surfaces, as illustrated in Scheme I.
 | ||
| Scheme 1 Heterolytic (A) and homolytic (B) pathways of the covalent aryldiazonium attachment at surfaces. | ||
The spontaneous grafting process has been mainly studied using p-substituted benzenediazonium salts on metals and carbon. The most popular reagent choice is 4-nitrobenzenediazonium tetrafluoroborate (4NBD) due to the fact that it is relatively stable, it is commercially available and it possesses a nitrophenyl group that can be used to electrochemically monitor the surface coverage via its electroreduction to phenylamine. Few reports however exist on the spontaneous grafting of diazonium salts with polynuclear aromatic backbones. Polynuclear aromatic molecules, such as biphenyls, azobenzenes, stilbenes, oligo(phenylenes), or anthracene, have been the subject of intense investigation due to their extended conjugated systems. Diazonium derivatives of these molecules have been used for fundamental studies of charge transfer through thin organic films and molecular aggregates,9,24–30 as well as for sensing applications31 in which the extended π-system and backbone rigidity are leveraged to allow fast electron transfer. With few exceptions, previous work on surface modifications using polynuclear aromatic diazonium derivatives has been almost exclusively carried out using an electrochemical approach. The spontaneous grafting of diazonium derivatives of polycyclic aromatic hydrocarbons (PAH–N2+), on the other hand, has not yet been investigated. This is surprising, since spontaneous grafting of aryldiazonium salts has been shown to lead to greater control over surface coverage and layer ordering,12 two properties that are of great importance for electronic and sensing applications.
In this work we report studies on the spontaneous grafting of diazonium derivatives of PAH compounds at carbon surfaces. We synthesized two diazonium salts that share a 1-nitronaphthalene backbone: 4-nitronaphthalenediazonium tetrafluoroborate (4NND) and 5-nitronaphthalenediazonium tetrafluoroborate (5NND); the structure of these two compounds is shown in Scheme II. We have used a combination of techniques in order to investigate their covalent attachment to carbon and compare it to that of their monocyclic analog 4NBD. The nitro group allowed us to monitor the grafting process and coverage in situ via infrared spectroscopy at the solid–liquid interface and ex situ via electrochemical methods. Thermal or solvolytic decomposition of aryldiazonium salts has been shown to play a role in the covalent grafting of aryldiazonium salts.32 Therefore, we studied the thermal stability and rate of solvolysis of 4NND and 5NND in order to understand whether these can affect their spontaneous attachment to carbon. We have found marked differences in the attachment rate of these two molecules and we discuss them in terms of their structural differences.
 | ||
| Scheme 2 Structures of 4-nitrobenzenediazonium tetrafluoroborate (4NBD), 4-nitronaphthalenediazonium tetrafluoroborate (4NND) and 5-nitronaphthalenediazonium tetrafluoroborate (5NND). | ||
Results and discussion
Grafting of 4NND and 5NND at carbon surfaces
The assembly of the diazonium salt derivatives of nitronaphthalene was first investigated ex situ. Carbon samples were immersed in 4NND and 5NND solutions, and thoroughly rinsed and dried prior to characterization via surface spectroscopy and electrochemical techniques. Fig. 1a shows the infrared reflection absorption spectroscopy (IRRAS) spectrum of an amorphous carbon (a-C) after modification in 1 × 10−3 M acetonitrile solutions of 4NND and 5NND for 1 h. The spectra show peaks at 1533 cm−1 and 1352 cm−1 that are assigned to the asymmetric and symmetric N–O stretching modes (νas(NO2) and νs(NO2)), respectively, of the nitro groups.33,34 The peaks at 767 and 790 cm−1 in the 4NND and 5NND traces, respectively, can be attributed to the C–H out of plane bending of the naphthalene backbone.33,34 No peaks could be detected in the 2300–2230 cm−1 region, where –N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N+ stretching modes would be expected (see the ESI†).34 The IRRAS spectroscopic signature of the 4NND and 5NND modified surfaces shown in Fig. 1a is in good agreement with that obtained from other nitroaromatic diazonium salts, such as 4-nitrobenezenediazonium tetrafluoroborate, and suggest that surface immobilization takes place spontaneously, as in the case of monocyclic aryldiazonium salts.35,36
N+ stretching modes would be expected (see the ESI†).34 The IRRAS spectroscopic signature of the 4NND and 5NND modified surfaces shown in Fig. 1a is in good agreement with that obtained from other nitroaromatic diazonium salts, such as 4-nitrobenezenediazonium tetrafluoroborate, and suggest that surface immobilization takes place spontaneously, as in the case of monocyclic aryldiazonium salts.35,36
 | ||
| Fig. 1 (a) IRRAS spectra of a-C substrates following immersion in 1 × 10−3 M 4NND and 5NND solutions for 1 h. (b) Cyclic voltammograms of 4NND and 5NND-modified a-C electrodes obtained in 0.1 M H2SO4 at 0.2 V s−1; the substrates were modified via immersion in 1 × 10−4 M aqueous solutions of 4NND and 5NND for 10 min. | ||
The grafting of nitronaphthalene diazonium salts on carbon was also investigated via electrochemistry using amorphous carbon electrodes. Fig. 1b shows the cyclic voltammetric response of 4NND and 5NND modified a-C electrodes in 0.1 M H2SO4 aqueous solutions. The modified a-C electrodes in Fig. 1b were prepared via spontaneous modification in 1 × 10−4 M aqueous solutions of the diazonium salts for 10 min. The cyclic voltammograms (CVs) show an irreversible cathodic peak at −0.87 V and −0.59 V (vs. Ag/AgCl) for 4NND and 5NND, respectively, that we attribute to the electroreduction of the nitroaryl groups.37 Subsequent potential sweeps do not display these peaks, which is consistent with a near complete reduction of the surface-bound ArNO2 groups. The difference of ∼0.28 V in the cathodic peak position for 4NND and 5NND suggests that the organic layers resulting from the spontaneous grafting of these two molecules over 10 min are different in structure and/or thickness. The cathodic peak for 4NND has a larger integrated area than that observed for 5NND. This result also suggests that a higher surface density of reducible nitroaryl groups is present at the carbon surface in the case of 4NND, which would be consistent with a thicker organic film. Both IRRAS and electrochemical results, however, indicate that both nitronaphthalene diazonium salts can be spontaneously grafted from solution onto amorphous carbon films.
In situ adsorption rate studies at carbon–solution interfaces
4NND and 5NND are positional isomers: in 4NND the –N2+ group is positioned on the same aromatic ring as the electron withdrawing –NO2 group, whereas in 5NND the –N2+ is positioned in the neighbouring aromatic ring of the nitronaphthalene backbone. In order to investigate whether this difference affected the spontaneous attachment of these molecules onto carbon surfaces we carried out in situ and ex situ studies of their reaction at the carbon surface.The in situ monitoring of the spontaneous grafting of 4NND and 5NND onto a-C substrates was carried out via attenuated total internal reflection Fourier transform infrared spectroscopy (ATR-FTIR) techniques as previously reported.35 Carbon coated trapezoids were used as ATR optical elements in a liquid cell containing water; at time zero, a 1 × 10−4 M aqueous solution of the diazonium salt was introduced into the cells and left to react at the carbon surface. Fig. 2a shows an example of ATR-FTIR spectra in the ν(NO2) spectral region measured at different times after the injection of the 4NND solutions into the liquid cell. Surface adsorption was monitored by plotting the net absorbance at the ∼1340 cm−1 peak maximum associated with the νs(NO2) mode; this peak was chosen over the asymmetric stretching mode because it was least affected by water vapour peaks across all of the ATR-FTIR datasets. The νs(NO2) net absorbance was corrected for any differences in pathlength by normalizing spectra against the same peak obtained from a standard KNO3 solution, thus obtaining curves of corrected net absorbance as a function of time. Fig. 2b shows adsorption curves obtained via this methodology for 4NND and 5NND; curves obtained for the monocyclic aromatic analog, 4NBD, previously investigated in our group35 are also reported for comparison purposes. In order to obtain curves that reflect the density of adsorbed molecules at the carbon surface as a function of time, we further normalized the curves in Fig. 2b by the relative absorption cross sections of the νs(NO2) mode for the three different molecules (see the ESI†). Fig. 2c shows the results of normalization by both pathlength and absorption cross sections; these curves display the changes in the surface density of the nitroaryl groups as a function of the deposition time obtained during the adsorption of 4NBD, 4NND and 5NND at the same carbon surface.
 | ||
| Fig. 2 (a) ATR-FTIR spectra collected as a function of time after injection of a 1 × 10−4 M 4NND aqueous solution into the liquid cell; spectra are offset for the sake of clarity. (b) Plot of the pathlength-corrected net absorbance of νs(NO2) as a function of deposition time obtained for 4NBD, 4NND and 5NND. (c) Plot of the νs(NO2) net absorbance normalized by pathlength and absorption cross-section (normalized net absorbance), as a function of deposition time. The normalized net absorbance is proportional to the surface density of the arylnitro groups at the carbon–solution interface. | ||
The deposition curves shown in Fig. 2c indicate that the rate of adsorption of the nitroaryl groups follows the order 4NBD > 4NND > 5NND. In all three cases two adsorption regimes can be observed: a fast first adsorption process takes place within the first 20 min followed by a slower adsorption process at longer times, in agreement with previous reports on 4NBD by Lehr et al.23 A first notable difference that emerges from Fig. 2c is that between the adsorption rate of 4NBD and 4NND. These two molecules share a p-nitrosubstituted benzenediazonium backbone, but differ in size due to the presence of an additional fused aromatic ring in 4NND. After the first 20 min of deposition, the average surface density of the arylnitro groups at the 4NBD modified carbon surfaces is equal to 3.4 times the density observed via modification with 4NND. This value is in excellent agreement with the previously observed surface density ratios of 2.7 obtained by Allongue et al. from electrografting experiments on the same diazonium salts, which were attributed to differences in the diazonium cation size.38 The similar surface density ratios obtained via electrografting and spontaneous adsorption suggest that differences in the adsorption rate between 4NBD and 4NND in Fig. 2c arise predominantly from differences in the molecular cross section.
A second significant difference is that observed between the two positional isomers, 4NND and 5NND. After the first 20 min of deposition, the average surface density of the nitroaryl groups at the 4NND modified carbon surfaces is equal to 3.1 times the density observed via modification with 5NND indicating that 4NND accumulates in significantly higher yields at a-C surfaces. 4NND and 5NND do not differ by their geometric molecular cross sections; therefore, the difference in the surface density shown in Fig. 2c must arise from the difference in the position of the –N2+ relative to the –NO2 group.
Ex situ studies of grafting rates at carbon surfaces
Ex situ data on the surface grafting of 4NND and 5NND at a-C substrates was obtained via electrochemical methods. Surface coverage (Γ) of the electroactive nitroaryl group was determined via integration of the reduction peaks35,38 after progressively longer immersion times. Fig. 3 shows Γ as a function of time for 1 × 10−4 M 4NND and 5NND on a-C substrates. The magnitude of Γ obtained from our experiments is in good agreement with literature reports of 15 × 10−10 mol cm−2 for the saturation coverage of 4NND at a flat surface.38 Results from ex situ characterisation are in agreement with those obtained via in situ spectroscopic monitoring at a-C surfaces: 4NND modified substrates show an increased surface density when compared to 5NND samples after the same deposition time. The difference in Γ values for 4NND and 5NND is consistent with the differences in the nitroaryl electroreduction potentials observed in Fig. 1 (∼0.28 V difference, see above), since the thicker 4NND layer can result in increased impedance to electron transfer at the organic–solution interface when compared to 5NND chemisorbed films. | ||
| Fig. 3 Surface coverage of nitroaryl groups (Γ) on a-C substrates as a function of deposition time in 1 × 10−4 M 4NND (○) and 5NND (■) solutions. | ||
The differences in the adsorption of the two positional isomers observed in situ, ultimately lead to different surface coverage in the chemisorbed organic layer, as shown by ex situ techniques. Since the geometric molecular cross section of 4NND and 5NND is similar, differences observed in adsorption rates must be attributed to differences in the position of the –N2+ relative to the –NO2 group. A change in the substitution position can have two important effects on these molecules: substitution position at C4 compared to C5 should lead to (a) higher diazonium salt stability towards dediazoniation and/or (b) changes in electron density, in particular at the diazo-substituted carbon. In order to understand whether one or both of these effects plays a role in determining the adsorption rate at the carbon–solution interface we carried out an investigation on the stability of 4NND and 5NND towards dediazoniation.
Dediazoniation of 4NND and 5NND salts
If the dediazoniation of aryldiazonium cations in solution was an important factor in determining the adsorption rates at the carbon surface, a significant difference in the dediazoniation rates in solution should also be observed. Based on the different positioning of the stabilizing nitro group with respect to the diazo group in 4NND and 5NND it is reasonable to expect differences in the stability and/or rate of dediazoniation. In order to test this hypothesis we investigated the thermal decomposition of 4NND and 5NND in the solid state and in solution.The stability towards thermal decomposition of 4NND and 5NND was first investigated in the solid state using TGA (thermogravimetric analysis) and DSC (differential scanning calorimetry). The weight percentage loss of both 4NND and 5NND upon heating under an inert atmosphere was measured for both compounds as illustrated in Fig. 4. The TGA results display an initial sharp weight loss between 120 °C and 160 °C for both compounds and then a gradual decline in weight as the temperature increased to 900 °C. The extrapolated onset temperatures of thermal decomposition were found to be 145 ± 2 °C and 126 ± 1 °C (90% confidence interval (C.I.)) for 4NND and 5NND, respectively. DSC data were used to calculate reaction enthalpys ΔRH associated with this decomposition process of −132.1 kJ mol−1 and −133.3 kJ mol−1 for 4NND and 5NND, respectively. Bräse et al.39 have proposed that the first step in the thermal decomposition of aryldiazonium tetrafluoroborate salts in the solid phase involves dediazoniation, in a process similar to a Schiemann reaction.40 Therefore, our results indicate that 5NND is more thermally labile than 4NND, as would be expected based on the stabilizing effect of the nitro group being closer to the diazo group in the case of 4NND.41
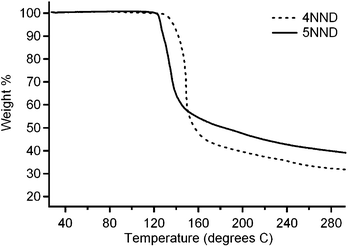 | ||
| Fig. 4 The thermogravimetric analysis of 4NND and 5NND under N2 in the range 0–900 °C at 10 °C min−1. | ||
In order to determine if any significant differences in dediazoniation rates were observed in solution, we carried out hydrolysis experiments using 4NND and 5NND. Aryldiazonium compounds hydrolyse via dediazoniation followed by formation of phenolic derivatives;1,42,43 this process can be followed via UV-vis by monitoring the disappearance of the absorption peak at ∼250 nm associated with the aryldiazonium groups, as previously reported.41,42,44Fig. 5a and 5b show the evolution of the absorption spectra of 1 × 10−4 M 4NND and 5NND solutions, respectively, as a function of time at 37 °C over 7 h: as hydrolysis of the aryldiazonium cation progresses, the absorption peaks at 259 nm (Fig. 5a) and 232 nm (Fig. 5b) decrease. Observed first-order hydrolysis rate constants were obtained by fitting the absorbance vs. time according to eqn (1) (shown in the insets in Fig. 5):41,45,46
 | (1) |
 | ||
| Fig. 5 UV-Vis spectra of 1 × 10−4 M solutions of 4NND (a) and 5NND (b) during hydrolysis at 37 °C over a period of 7 h. The insets show the corresponding first-order kinetic plots obtained from the UV-vis data (see eqn (1)). | ||
Our results indicate that, despite showing differences in thermal stability in the solid phase, 4NND and 5NND are equally stable towards dediazoniation under typical conditions used for spontaneous adsorption at surfaces. The remarkable difference in the adsorption rate observed in Fig. 2 and Fig. 3 therefore cannot be attributed to dediazoniation reactions that take place in solution but must arise from differences in the interaction of 4NND and 5NND at the carbon surface due to the differences in electron density between the two positional isomers.
Therefore, electrochemical measurements were carried out to find the reduction potential of the aryldiazonium functional group. The peak potential at which the reduction peak is observed shifts with the position of acceptor levels in solution. A cathodic sweep in the 7 × 10−4 M solutions of 4NND and 5NND in anhydrous acetonitrile shows an irreversible cathodic wave with Ep at 0.05 ± 0.01 V and −0.20 ± 0.02 V (vs. Saturated Calomel Electrode C.I. = 90%) for 4NND and 5NND, respectively (see ESI†). The more negative potential obtained for 5NND indicates that its reduction is more difficult to achieve than that of its positional isomer 4NND.
Conclusions
The spontaneous grafting of fused aromatic nitro compounds onto amorphous carbon surfaces was investigated using a combination of spectroscopic and electrochemical techniques. The in situ grafting of 4NND and 5NND onto amorphous carbon surfaces was monitored and compared with that of their monocyclic analog 4NBD. Differences in the adsorption rate and the final surface coverage between 4NBD and 4NND could be explained based on geometric molecular cross-sections, however, the remarkable difference in the adsorption and chemisorption rates between 4NND and 5NND could not be explained via steric arguments.The solvolysis of 4NND and 5NND was found to occur over similar timescales under the experimental conditions used for the spontaneous attachment of these two aryldiazonium salts, therefore, the differences in the coverage cannot be ascribed to the faster degradation in solution of one of the positional isomers. Adsorption rates, on the other hand, appear to correlate very well with the reduction potential of the aryldiazonium cation, which was found to be more positive for 4NND compared to 5NND. These results are in agreement with reactivity trends observed for monocyclic aryldiazonium salts, which suggest that cations with lower lying electron-acceptor levels are more easily grafted than electron rich cations.
This work shows, for the first time, the spontaneous grafting of diazonium derivatives of n-PAHs onto amorphous carbon substrates. Our results indicate that the spontaneous grafting of diazonium derivatives of n-PAHs can lead to different layers at carbon surfaces, depending on the position of the –N2+ functional group with respect to the other substituents in the condensed ring system. This work presents a clear example of positional isomers displaying very different grafting behaviours and highlights the importance of substituent position which may be useful when choosing compounds for sensing and electronic applications.
Experimental
Chemicals and materials
Acetonitrile (ACN, Fisher, HPLC grade), anhydrous acetonitrile (99.8%, Sigma) sulphuric acid (95–97%, Sigma) and fluoroboric acid (48% in H2O, Sigma) were used without further purification. All other solvents were reagent grade and used as received. Ammonium chloride (≥99.5%), hydroxylamine hydrochloride (>98%), potassium hydroxide (pellets, >85%), potassium nitrate (≥98%), sodium nitrite (≥99%), tetrabutylammonium perchlorate (≥99%), 1-nitronaphthalene (99%), 1, 5-dinitronaphthalene (≥97%) and 4-nitrobenzenediazonium tetrafluoroborate (4NBD, 97%) were all obtained from Sigma and used as received. Sodium sulphide hydrate and ammonia hydroxide solution (35%) were sourced from Riedel-de Haen and AnalaR, respectively. Amorphous carbon substrates were prepared via DC magnetron sputtering as previously described,35 and consisted of a 70 nm carbon layer with a 300 nm thick titanium underlayer.47 Structural characterization of these films has previously shown that they are highly graphitic with an sp2 content of approximately 85%.35 Glassy carbon substrates were purchased from SPI (Glas 11), polished using 0.3 μm alumina slurry and rinsed in acid and in excess water prior to our experiments.4-Nitro-1-naphthylamine
4-Nitro-1-naphthylamine was prepared according to reported literature procedures starting from 1-nitronaphthalene.48 1-Nitronaphthalene (1.00 g, 6.0 mmol) and hydroxylamine hydrochloride (2.50 g, 3.6 mmol) were dissolved in 60 mL of ethanol in a 250 mL flask. While maintaining the temperature between 50 °C and 60 °C, a filtered solution of potassium hydroxide (5.00 g, 8.9 mmol) dissolved in 32 mL methanol was added gradually with mechanical stirring over a period of 45 min. After an additional hour of vigorous stirring the solution was poured into 350 mL of cold water. The resulting precipitate was filtered by suction and washed with water. Purification by recrystallisation in ethanol and drying under high vacuum results in 4-nitro-1-naphthylamine as long golden orange needles (0.77 g, 77%); νmax (thin film) 3401, 1515, 1311, 824, 777 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.01 (d, 1H, J = 8.5 Hz, Ar–H), 8.41 (d, 1H, J = 8.5 Hz, Ar–H), 7.86 (d, 1H, J = 8.5 Hz, Ar–H), 7.75 (t, 1H, J = 8.5 Hz, Ar–H), 7.59 (t, 1H, J = 8.5 Hz, Ar–H), 6.71 (d, 1H, J = 8.5 Hz, Ar–H), 4.96 (br s, 2H, NH2); 13C NMR (400 MHz, CDCl3) δ 149.0 (q), 137.0 (q), 130.0, 128.7, 127.7 (q), 126.0, 124.9, 121.8 (q), 121.0, 106.4; m/z HRMS (EI+) Calcd. for C10H8N2O2 [M]+ 188.0586. Found 188.0596.4-Nitro-naphthalenediazonium tetrafluoroborate (4NND)
4NND was prepared from 4-nitro-1-naphthylamine according to reported literature procedures.49 4-Nitro-1-naphthylamine (0.20 g, 1.0 mmol) was dissolved in tetrafluoroboric acid (0.18 g, 2.0 mmol). Additional tetrafluoroboric acid (approx. 5 mL) was used in order to fully dissolve the starting material. Following cooling to −5 °C, a 1 × 10−3 M solution of sodium nitrite in water was added dropwise over 30 min. The precipitate obtained was filtered by suction, washed with an ice cold ether–methanol mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and cold ethanol, and dried under high vacuum yielding the diazonium salt as a dark yellow solid (0.19 g, 95%); νmax (thin film) 2275, 1538, 1505, 1356 cm−1; 1H NMR (600 MHz, d-MeOH) δ 9.34 (d, 1H, J = 8.5, Ar–H), 8.64 (d, 1H, J = 8.5, Ar–H), 8.57 (d, 1H, J = 8.5, Ar–H), 8.53 (d, 1H, J = 8.5, Ar–H), 8.29 (t, 1H, J = 8.5, Ar–H), 8.19 (t, 1H, J = 8.5, Ar–H); 13C NMR (600 MHz, d-MeOH) δ 136.9, 134.0, 132.9, 129.7 (q), 125.0 (q), 125.0, 122.7, 122.0, 115.0 (q), 115.0 (q).
:1) and cold ethanol, and dried under high vacuum yielding the diazonium salt as a dark yellow solid (0.19 g, 95%); νmax (thin film) 2275, 1538, 1505, 1356 cm−1; 1H NMR (600 MHz, d-MeOH) δ 9.34 (d, 1H, J = 8.5, Ar–H), 8.64 (d, 1H, J = 8.5, Ar–H), 8.57 (d, 1H, J = 8.5, Ar–H), 8.53 (d, 1H, J = 8.5, Ar–H), 8.29 (t, 1H, J = 8.5, Ar–H), 8.19 (t, 1H, J = 8.5, Ar–H); 13C NMR (600 MHz, d-MeOH) δ 136.9, 134.0, 132.9, 129.7 (q), 125.0 (q), 125.0, 122.7, 122.0, 115.0 (q), 115.0 (q).
5-Nitronaphthalen-l-amine
5-Nitro-1-naphthalen-1-amine was prepared according to reported literature procedures starting from 1,5-dinitronaphthalene.50 A solution of ammonium chloride (2.80 g, 53 mmol) in concentrated aqueous ammonia was added to a suspension of 1,5-dinitronaphthalene (1.60 g, 7.0 mmol) in water (22 mL). The solution was heated at 80–85 °C with mechanical stirring. Sodium sulphide (1.91 g, 24 mmol) was added in three separate portions over 30 min. The mixture was heated for 4 h at 85 °C and filtered through a heated Büchner funnel. The solid was extracted twice with dichloromethane and dried over anhydrous magnesium sulphate. Excess solvent was removed and the residual material was purified by silica gel column chromatography. Elution with hexane–ethyl acetate (4:1), removal of solvent and drying under high vacuum results in 5-nitronaphthalen-1-amine as a brown-red solid (0.38 g, 40%); νmax (thin film) 3414, 3335, 3238, 1509, 1316, 1278, 773 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.15 (d, 2H, J = 7.5 Hz), 7.92 (d, 1H, J = 8.7 Hz), 7.52 (m, 2H), 6.93 (d, 1H, J = 7.5 Hz), 4.29 (br s, 2H, NH2); 13C NMR (400 MHz, CDCl3) δ 147.5 (q), 142.7 (q), 130.0, 127.0, 126.1 (q), 124.8 (q), 123.6, 122.8, 113.5, 111.3; m/z HRMS (EI+) Calcd. for C10H8N2O2 [M]+ 188.0586. Found [M]+H 189.0662.
5-Nitro-naphthalenediazonium tetrafluoroborate (5NND)
5NND was prepared from 5-nitro-1-naphthylamine (0.19 g, 1.0 mmol) according to reported literature procedures.49 Additional tetrafluoroboric acid (approx. 5 mL) and water (approx. 5 mL) was used in order to fully dissolve the starting material. The precipitate obtained was filtered by suction, washed with an ice cold ether–methanol mixture (4:1) and cold ethanol, and dried under high vacuum yielding the diazonium salt as a light brown solid (0.13 g, 75%); νmax (thin film) 2276, 1531, 1348, 800, 755 cm−1; 1H NMR (600 MHz, d-MeOH) δ 9.39 (d, 1H, J = 8.5 Hz), 9.29 (d, 1H, J = 8.0 Hz), 8.81 (d, 1H, J = 8.5 Hz), 8.70 (d, 1H, J = 8.0 Hz), 8.27 (m, 2H); 13C NMR (600 MHz, d-MeOH) δ 148.2 (q), 138.4, 138.3, 131.8, 129.1, 128.6 (q), 127.7, 127.6, 126.6 (q), 112.8 (q).
Sample characterization
Infrared reflection absorption spectroscopy (IRRAS) was carried out on a Bruker Tensor 27 Fourier transform infrared (FTIR) spectrometer, equipped with a mercury-cadmium-telluride (MCT) detector, using a VeeMaxII variable angle specular reflectance accessory with a wire grid polarizer. All IRRAS measurements were obtained using p-polarized light at an 80° angle of incidence from the surface normal. 256 Scans with a resolution of 4 cm−1 were collected for each sample and a bare a-C substrate was used as the background. In situ monitoring of the spontaneous grafting of 4NBD, 4NND and 5NND onto carbon substrates was carried out using a custom-built ATR cell for in situ monitoring.35 The internal reflection element was a trapezoid made from 1245 μm thick, double side polished, undoped silicon wafers (Virginia Semiconductors, resistivity >100 Ω cm) with polished 45° edges. The trapezoid was sputter coated on its longest face with amorphous carbon films and a polydimethylsiloxane (PDMS) gasket was used to seal the carbon side of the trapezoid forming a liquid cell. Background spectra were collected via the injection of deionized water; afterwards, a degassed solution of the aryldiazonium in deionized water was injected into the cell. Spectra were collected at regular time intervals after injection; 100 scans at 4 cm−1 resolution were averaged for each time point over the course of 3 h. Upon completion of the grafting experiment a 0.2 M aqueous KNO3 solution was injected into the cell; the nitrate peak at 1338 cm−1 from this spectrum was used to normalize all spectra in order to correct for differences in the optical pathlength. In order to compare grafting rates for the different compounds an internal standard method was used to normalize for differences in the cross section among 4NBD, 4NND and 5NND.51Thermogravimetric analysis (TGA) was performed on a Pyris 1 TGA (Perkin Elmer). Measurements were recorded under N2 atmosphere in the range 0–900 °C at 10 °C min−1. Differential scanning calorimetry (DSC) was carried out on a Diamond DSC (Perkin Elmer). These measurements were collected in the range 0–200 °C at 5 °C min−1 and in the range 90–180 °C at 0.5 °C min−1. UV-Vis was carried out on a Lambda35 (Perkin Elmer) spectrometer equipped with a thermostated cell; spectra were collected at 1 nm resolution in the range 220–470 nm at 37 °C, at regular time intervals.
Aryldiazonium grafting on a-C substrates was carried out by immersing the samples in aqueous solutions of 4NND and 5NND for varying lengths of time. After grafting, the substrates were washed and sonicated in water and dried under argon prior to electrochemical analysis. Cyclic voltammetry was carried out on a CHI660C potentiostat using Pt wire and Ag/AgCl (IJ Cambria) as counter and reference electrodes, respectively. Experiments were carried out in a previously described Teflon cell52 with an electrode area of 0.067 cm2. Electrochemical analysis was performed at room temperature in argon purged 0.1 M H2SO4 solutions. Electrochemistry of aryldiazonium salts in non-aqueous solutions was carried out in anhydrous acetonitrile using 0.1 M tetrabutylammonium perchlorate (NBu4ClO4) as supporting electrolyte;6 potentials were measured against a Pt pseudo-reference electrode and corrected using ferrocene as a standard.
Acknowledgements
This publication has emanated from research conducted with the financial support of Science Foundation Ireland under grant number 09/RFP/CAP2174. The authors are grateful to Dr Michael E.G. Lyons for granting access to instrumentation.References
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure., John Wiley & Sons, Inc, New York, 4th edn, 1992 Search PubMed.
- K. Kikukawa and T. Matsuda, Chem. Lett., 1977, 159–162 CrossRef CAS.
- D. Belanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- S. Mahouche-Chergui, S. Gam-Derouich, C. Mangeney and M. M. Chehimi, Chem. Soc. Rev., 2011, 40, 4143–4166 RSC.
- E. Ahlberg, B. Helgee and V. D. Parker, Acta Chem. Scand., Ser. B, 1980, 34b, 181–186 CrossRef.
- M. Delamar, R. Hitmi, J. Pinson and J. M. Savéant, J. Am. Chem. Soc., 1992, 114, 5883–5884 CrossRef CAS.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- J. L. Bahr and J. M. Tour, Chem. Mater., 2001, 13, 3823–3824 CrossRef CAS.
- F. R. F. Fan, J. P. Yang, L. T. Cai, D. W. Price, S. M. Dirk, D. V. Kosynkin, Y. X. Yao, A. M. Rawlett, J. M. Tour and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 5550–5560 CrossRef CAS.
- M. P. Stewart, F. Maya, D. V. Kosynkin, S. M. Dirk, J. J. Stapleton, C. L. McGuiness, D. L. Allara and J. M. Tour, J. Am. Chem. Soc., 2004, 126, 370–378 CrossRef CAS.
- A. Adenier, E. Cabet-Deliry, A. Chaussé, S. Griveau, F. Mercier, J. Pinson and C. Vautrin-Ul, Chem. Mater., 2005, 17, 491–501 CrossRef CAS.
- F. Barriere and A. J. Downard, J. Solid State Electrochem., 2008, 12, 1231–1244 CrossRef CAS.
- D. Ghosh and S. W. Chen, Chem. Phys. Lett., 2008, 465, 115–119 CrossRef CAS.
- D. Ghosh, S. Pradhan, W. Chen and S. W. Chen, Chem. Mater., 2008, 20, 1248–1250 CrossRef CAS.
- N. Griffete, F. Herbst, J. Pinson, S. Ammar and C. Mangeney, J. Am. Chem. Soc., 2011, 133, 1646–1649 CrossRef CAS.
- L. Laurentius, S. R. Stoyanov, S. Gusarov, A. Kovalenko, R. B. Du, G. P. Lopinski and M. T. McDermott, ACS Nano, 2011, 5, 4219–4227 CrossRef CAS.
- F. Mirkhalaf, J. Paprotny and D. J. Schiffrin, J. Am. Chem. Soc., 2006, 128, 7400–7401 CrossRef CAS.
- A. Adenier, N. Barre, E. Cabet-Deliry, A. Chausse, S. Griveau, F. Mercier, J. Pinson and C. Vautrin-Ul, Surf. Sci., 2006, 600, 4801–4812 CrossRef CAS.
- F. Le Floch, J.-P. Simonato and G. Bidan, Electrochim. Acta, 2009, 54, 3078–3085 CrossRef CAS.
- J. -M. Seinberg, M. Kullapere, U. Mäeorg, F. C. Maschion, G. Maia, D. J. Schiffrin and K. Tammeveski, J. Electroanal. Chem., 2008, 624, 151–160 CrossRef CAS.
- P. Abiman, G. G. Wildgoose and R. G. Compton, J. Phys. Org. Chem., 2008, 21, 433–439 CrossRef CAS.
- M. Toupin and D. Belanger, Langmuir, 2008, 24, 1910–1917 CrossRef CAS.
- J. Lehr, B. E. Williamson and A. J. Downard, J. Phys. Chem. C, 2011, 115, 6629–6634 CAS.
- A. P. Bonifas and R. L. McCreery, Nat. Nanotechnol., 2010, 5, 612–617 CrossRef CAS.
- A. J. Bergren, K. D. Harris, F. Deng and R. L. McCreery, J. Phys.: Condens. Matter, 2008, 20, 374117 CrossRef.
- J. Ru, B. Szeto, A. Bonifas and R. L. McCreery, ACS Appl. Mater. Interfaces, 2010, 2, 3693–3701 CAS.
- S. Ranganathan, I. Steidel, F. Anariba and R. L. McCreery, Nano Lett., 2001, 1, 491–494 CrossRef CAS.
- D. K. James and J. M. Tour, Chem. Mater., 2004, 16, 4423–4435 CrossRef CAS.
- H. Jian and J. M. Tour, J. Org. Chem., 2005, 70, 3396–3424 CrossRef CAS.
- J. He, B. Chen, A. K. Flatt, J. J. Stephenson, C. D. Doyle and J. M. Tour, Nat. Mater., 2006, 5, 63–68 CrossRef CAS.
- C. F. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710–1712 RSC.
- J. Agullo, S. Canesi, F. Schaper, M. Morin and D. Bèlanger, Langmuir, 2012, 28, 4889–4895 CrossRef CAS.
- V. Librando and A. Alparone, J. Hazard. Mater., 2008, 154, 1158–1165 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley & Sons, Chichester, 3rd edn, 2001 Search PubMed.
- R. J. Cullen, D. Jayasundara, L. Soldi, J. Cheng, G. DuFaure and P. E. Colavita, Chem. Mater., 2012, 24, 1031–1040 CrossRef CAS.
- D. R. Jayasundara, R. J. Cullen, L. Soldi and P. E. Colavita, Langmuir, 2011, 27, 13029–13036 CrossRef CAS.
- P. A. Brooksby and A. J. Downard, Langmuir, 2004, 20, 5038–5045 CrossRef CAS.
- P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- S. Bräse, S. Dahmen, C. Popescu, M. Schroen and F.-J. Wortmann, Chem.–Eur. J., 2004, 10, 5285–5296 CrossRef.
- G. Balz and G. Schiemann, Ber. Dtsch. Chem. Ges., 1927, 60, 1186–1190 CrossRef.
- P. S. J. Canning, K. McCrudden, H. Maskill, B. Sexton and J. Chem. Soc, J. Chem. Soc., Perkin Trans. 2, 1999, 2735–2740 RSC.
- B. Quintero, M. C. Cabeza, M. I. Martínez, P. Gutiérrez and P. J. Martínez, Can. J. Chem., 2003, 81, 832–839 CrossRef CAS.
- C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS.
- B. Quintero, M. I. Martínez Puentedura, M. T. Megías, M. C. Cabeza, M. P. Gutiérrez and P. J. Martínez de las Parras, J. Chromatogr., A, 2004, 1035, 227–236 CrossRef CAS.
- R. Pazo-Llorente, H. Maskill, C. Bravo-Diaz and E. Gonzalez-Romero, Eur. J. Org. Chem., 2006, 2201–2209 CrossRef CAS.
- R. Pazo-Llorente, M. J. Sarabia-Rodriguez, C. Bravo-Diaz and E. Gonzalez-Romero, Int. J. Chem. Kinet., 1999, 31, 73–82 CrossRef CAS.
- B. Sun, P. E. Colavita, H. Kim, M. Lockett, M. S. Marcus, L. M. Smith and R. J. Hamers, Langmuir, 2006, 22, 9598–9605 CrossRef CAS.
- C. C. Price and S.-T. Voong, Org. Synth., 1948, 28, 80 CAS.
- A. Hermans, A. T. Seipel, C. E. Miller and R. M. Wightman, Langmuir, 2006, 22, 1964–1969 CrossRef CAS.
- J. G. Rodríguez and J. L. Tejedor, J. Org. Chem., 2002, 67, 7631–7640 CrossRef.
- M. K. Bellamy, J. Chem. Educ., 2010, 87, 1399–1401 CrossRef CAS.
- L. Soldi, R. J. Cullen, D. R. Jayasundara, E. M. Scanlan, S. Giordani and P. E. Colavita, J. Phys. Chem. C, 2011, 115, 10196–10204 CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: details of the normalisation procedure for the infrared absorption cross sections for the three compounds used in our experiments, cyclic voltammetry of 4NND and 5NND in acetonitrile and additional infrared spectra of the organic films. See DOI:10.1039/c2ra20292a/ |
| This journal is © The Royal Society of Chemistry 2012 |