DOI:
10.1039/C2RA01347A
(Paper)
RSC Adv., 2012,
2, 6535-6541
Activation of serpentine for CO2 mineralization by flux extraction of soluble magnesium salts using ammonium sulfate†
Received
22nd December 2011
, Accepted 21st March 2012
First published on 22nd June 2012
Abstract
This paper concerns the growing role of cheap and potentially recyclable ammonium salts in CO2 mineralization. The powerful hyphenated technique TG-FTIR, along with XRD and ICP-AES, were used to shed light on the underlying chemistry and measure the efficiency of magnesium ion extraction from a Finnish serpentinite in contact with molten ammonium sulfate above 300 °C. From micro- and gram-scale tests, flux extraction as epsomite [MgSO4·7H2O] proceeds via the intermediacy of Tutton salts, NH4/Mg double sulfates increasingly rich in Mg. Extraction is effected through the agency of acidic derivatives, principally ammonium bisulfate and sulfamic acid, which are created sequentially from ammonium sulfate in situ. However, sulfamic acid volatilizes and/or decomposes at a significant rate by 400 °C. This loss mechanism is primarily responsible for the modest recovery of Mg (50–60%). An improved process would operate below 400 °C where Mg extraction as efremovite [(NH4)2Mg2(SO4)3] is effective. Future experiments evaluating the use of humid air to stabilize the bisulfate and impede the loss of flux are recommended.
1. Introduction
Many scientists are now convinced that the trend of increasing frequency and severity of what are considered “normal” (seasonal) climatic and meteorological disturbances, e.g., flooding and destruction from typhoons and hurricanes, melting of polar ice, etc., are but portents of worse to come under a “business-as-usual” energy scenario. Global warming due to rising anthropogenic emissions of carbon dioxide is leading inexorably towards a “tipping point”, beyond which irreversible climate change may result with potentially disastrous consequences. While some valuable trends in better carbon management of the energy sector are afoot, e.g., more efficient energy use, reduction in carbon intensity of fossil fuels, phasing-in of renewable (carbon-neutral) energy cycles, etc., their climatic impact will probably not register for decades.1 Recourse to more direct and immediate measures to stabilize or reduce atmospheric levels of CO2 is now inevitable. Efficient and affordable carbon capture and storage (CCS) technologies must be developed and implemented on a massive scale. The primary strategy is interception of CO2 emissions at source for all large scale utilities, e.g., power stations, cement factories, etc., where its relatively concentrated form makes scrubbing more economic. In the longer term, miniature absorbers for direct trapping of CO2 from the atmosphere, provided they are distributed in literally huge numbers, may eventually also contribute tangibly to climate stabilization. Proof-of-principle for such “artificial trees” is now well established.2–4 By way of permanent storage/disposal, mineral carbonation offers the most guaranteed option on the requisite scale.5,6 In effect, exposure of ultramafic rocks rich in alkaline earths (Mg, Ca), e.g., peridotites, serpentines, olivines, etc., to CO2 under conditions promoting rapid absorption constitutes a hyper-accelerated version of the natural process of “weathering” and should be ecologically benign. The carbonate products are stable and require no post-process monitoring (of leakage), unlike geological storage via subterranean gas- or supercritical fluid-injection.7 The world capacity for CO2 sequestration as mineral carbonates is estimated in the 104–106 gigaton carbon (GtC) range. This exceeds the estimated level of all known fossil fuel reserves (5 × 103 GtC), and dwarfs all other disposal routes combined, i.e., ocean, deep saline, depleted oil and gas reservoirs, enhanced oil recovery, etc. (∼103 GtC). Unfortunately, mineral carbonation suffers from one serious drawback. The kinetics for direct gas–solid carbonation of the intact mineral are too slow, and current research is attempting to realize an economic means of mineral activation. After the pioneering exploratory work of Lackner and co-workers in the mid-1990s,8,9 the direct (gas–solid) approach was all but abandoned or superseded by developments in indirect or “wet” methods. At present, the most successful of these is generally agreed to be one-pot salt-assisted aqueous slurry processing at 180 °C under supercritical CO2.10 This last constraint, indicative of mass transfer limitations, is not surprising since the solubility of CO2 in the acidic environment necessary for mineral dissolution at practical rates is very low. Thus, it suffers from chemically conflicting requirements, and developments under consideration to circumvent this limitation, such as pH swing,11,12 introduce greater complexity and cost into the process. Rates are also impeded by the build-up of a passivating layer of SiO2.13 Nevertheless, most current research favours the “pre-dissolution” approach.14–22 Recently, Åbo Akademi University (ÅAU) has been developing a staged process which may offer certain advantages, as shown schematically in Fig. 1. First, the Mg component is extracted from serpentine using ammonium sulfate (AS) as a cheap and potentially recyclable flux:23–25| | | Mg3Si2O5(OH)4 + 3(NH4)2SO4 → 3[Mg2+SO42−] + 2SiO2 + 5H2O + 6NH3 | (1) |
during which NH3 is trapped out separately as ammonium hydroxide for subsequent use. In Stage 2, an aqueous wash leaves behind the solid residue (SiO2), and soluble Mg2+ ions are precipitated as ultra-fine brucite by controlled addition of NH4OH created in situ:| | | 3[Mg2+SO42−] + 6NH4OH → 3Mg(OH)2 + 6NH4+ + 3SO42− | (2) |
after which AS is crystallized out of the mother liquor for recycling to Stage 1. Brucite is then carbonated in a pressurized fluidized bed reactor:| | | 3Mg(OH)2 + 3CO2 → 3MgCO3 + 3H2O | (3) |
in which heat from this exothermic process (Stage 3) can, in principle, be recycled to the energy-demanding flux extraction (Stage 1). The overall balance for serpentine carbonation is then:| | | Mg3Si2O5(OH)4 + 3CO2 → 3MgCO3 + 2SiO2 + 2H2O | (4) |
![Scheme of staged CO2 mineralization process based on Mg pre-extraction via ammonium sulfate as recyclable flux [Åbo Akademi University].](/image/article/2012/RA/c2ra01347a/c2ra01347a-f1.gif) |
| | Fig. 1 Scheme of staged CO2 mineralization process based on Mg pre-extraction via ammonium sulfate as recyclable flux [Åbo Akademi University]. | |
Under standard conditions, reaction (4) is actually mildly exothermic (ΔH0 ≈ −35 kJ mol−1 Mg).8,26 In recompense for the added complexity of this staged (precipitation) process, it could provide by-products in high purity and having market value, e.g., iron oxides for the iron & steel industry. The carbonated products, magnesite and hydromagnesite [more familiarly known as “basic” magnesium carbonate, 4MgCO3·Mg(OH)2·4H2O], are commodities in their own right, and substantial recent interest is being shown in these as smoke- and fire-retardants.27,28 An outlet specific for Singapore from large-scale mineral processing would be land reclamation.29,30 Environmental benefits may also accrue from their use. For example, magnesite is reported to have potential for heavy-metal ion sequestration.31,32 However, the associated chemistry, heat and material cycles must be well-understood and optimized. Efficiency limitations already recognized and under study include inadequate heat recycling (energy loss) from carbonation (Stage 3) to the flux extraction (Stage 1), as well as a substantial energy penalty associated with recovery of solid ammonium sulfate after brucite precipitation (Stage 2), even from concentrated aqueous solution.33,34
This paper focuses on the underlying chemistry of flux extraction, identifies a range of solid-state intermediate structures in the product, reports typical yields of Mg from gram-scale extractions, identifies loss mechanisms that currently detract from efficient recycling of ammonium sulfate, and offers potential directions towards a more effective process. A related paper, to be published elsewhere,35 summarizes progress in gas–solid carbonation (Stage 3 of the cycle) in our laboratories, focusing on the promoting effect of low levels of steam on the kinetics of both brucite and magnesia conversion and their possible inter-relationship.36 A companion paper in this journal37 reports on the role of ammonium salts in mechanochemical processing as an effective means for Mg extraction and carbonation under ambient conditions.
2. Experimental
2.1 Materials
The serpentinite was received in lump form from a nickel mine in Hitura, Finland, and milled briefly to <45 μm (#325 mesh). In XRD, it showed a diffraction pattern fitting best to the hexagonal lizardite‡polymorph [(Mg,Al)3(Si,Fe)2O5(OH)4; #00-050-1625].
Weak additional lines of magnetite [Fe3O4; #03-065-3107] were also present. XRF analysis showed the serpentinite to be of high purity, having a Mg content (as the oxide) of 43 wt% [theor. = 43.6%]. The BET surface area [N2 physisorption, 77 K] was measured as ∼30 m2 g−1, indicating the presence of substantial porosity. All other chemicals were reagent grade (>99%, Aldrich) and used as supplied. The magnesia (MgO, #325 mesh) had a BET surface area of 204 m2 g−1.
2.2 Methodology
2.2.1 Gram-scale flux extraction.
Lizardite–ammonium sulfate (AS)§ mixtures (1 g total) in various proportions were weighed into a 7 × 1 cm ceramic boat and slid into the central isothermal zone of a sealable horizontal tubular (75 × 3.5 cm od) quartz furnace (Lenton Thermal Solutions) via a custom-built rail-mounted assembly containing a hooked tungsten rod to allow easy loading and withdrawal. Samples were heated at 10 °C min−1 to the target temperature and held isothermal for 1 h under dry air flow (150 ml min−1). The discharged product was recovered and weighed. Control studies on MgO were conducted similarly.
2.3 Analytical techniques
2.3.1 TG-FTIR.
Thermogravimetry (TG) coupled to Fourier-transform infrared (FTIR) spectroscopy is a powerful tool in the characterization of solid materials. A controlled heating ramp is applied to a sample suspended on a sensitive microbalance under carrier gas flow and the weight change recorded. Any infrared-active evolved gas products are continuously swept into the IR spectrometer, identified and monitored by transmission in a suitable optical cell. In this laboratory, a Setaram Setsys 12 TG unit is linked to a Digilab Excalibur series FTIR spectrometer fitted with a KBr beamsplitter and a mid-range (liquid N2-cooled) MCT detector. The interface unit comprises a transfer line and 10 cm pathlength gas cell, both thermostatted at 220 °C. In a typical experiment, 40–50 mg of sample was loaded in an alumina crucible in dry air (30 ml min−1, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 purge:auxiliary flow) and the furnace ramped at 10 °C min−1 to a target temperature and held isothermal for 1 h. Time-resolved FTIR data acquisition was made using the kinetics mode, in which a spectrum was recorded every 20 s, representing an average of 60 co-added scans at 4 cm−1 resolution. Evolution profiles specific to H2O vapour (band centre at 3900 cm−1), CO2, (2340 cm−1) and NH3 (946 cm−1) were tracked using pre-existing spectral “markers”, while SO2 (1360 cm−1) and N2O (2220 cm−1) were initially identified from the EPA gas-phase spectral library during preliminary runs. For humid experiments, water vapour was supplied from a Setaram Wetsys unit set to deliver 50% RH at 50 °C. This corresponded to a pressure of ∼6 kPa, giving 3 kPa at the sample, or 3 vol%.
:1 purge:auxiliary flow) and the furnace ramped at 10 °C min−1 to a target temperature and held isothermal for 1 h. Time-resolved FTIR data acquisition was made using the kinetics mode, in which a spectrum was recorded every 20 s, representing an average of 60 co-added scans at 4 cm−1 resolution. Evolution profiles specific to H2O vapour (band centre at 3900 cm−1), CO2, (2340 cm−1) and NH3 (946 cm−1) were tracked using pre-existing spectral “markers”, while SO2 (1360 cm−1) and N2O (2220 cm−1) were initially identified from the EPA gas-phase spectral library during preliminary runs. For humid experiments, water vapour was supplied from a Setaram Wetsys unit set to deliver 50% RH at 50 °C. This corresponded to a pressure of ∼6 kPa, giving 3 kPa at the sample, or 3 vol%.
2.2.2 XRD.
X-Ray patterns were obtained from lightly pressed powder samples on a Bruker D8 Powder Diffractometer with Cu–Kα source (λ = 1.5418 Å) over the range 2θ = 10−80° at a step size of 0.008°.
2.2.3 ICP-OES.
Aqueous extracts of gram-scale flux extraction, obtained from 50 ml suspensions (∼200 mg sample) suitably filtered and diluted (×100) in DI water, were analyzed for elemental Mg and S (1–10 ppm calibrated range) in a Varian Vista-MPX inductively-coupled plasma optical emission spectrometer.
3. Results and discussion
3.1 Microscale studies of flux extraction by TG-FTIR
3.1a Thermolysis of ammonium sulfate.
A control experiment by TG-FTIR on pure ammonium sulfate (AS) under dry air up to 480 °C is shown in Fig. 2. The onset of degradation is at ∼250 °C with NH3 and H2O evolution peaking at ∼300 °C. The corresponding weight loss up to 350 °C was ∼25%, suggesting that sulfamic acid (SA) is an intermediate:| | | (NH4)2SO4 → NH3SO3 + NH3 + H2O (Δm = −26.5%) | (5) |
 |
| | Fig. 2 Thermogram of ammonium sulfate up to 480 °C by TG-FTIR. | |
The entirety of the sample was eventually lost by ∼450 °C, accompanied by an SO2 peak at 400 °C, N2O (not shown), along with a second stage of water loss. A control study on SA essentially reproduced the above features (see Fig. S2, ESI†), confirming its formation in situ from AS and ultimate decomposition, possibly via hydroxylamine:
| | | 2NH3SO3 → 2SO2 [+2NH2OH → N2O + H2O + 2H2] | (6) |
Fig. S2 (ESI†) also shows initial weight loss suggestive of “boil off” in the superheated liquid state (SA melts at 205 °C38), as little evolved gas was detected below 300 °C. This explains the observed pressure build-up in the TG furnace from both AS and SA, which was eventually traced to partial blockage by re-solidified material in the vent line. Although the sublimate was successfully rinsed away with water between experiments, both effects (sublimation and irreversible decomposition) constitute losses that must be scrupulously avoided to maintain flux recycling at high efficiency.
As ammonium bisulfate (ABS) is almost certainly the primary intermediate, formed by simple NH3 loss from AS, eqn. (5) can be subdivided into two separate steps:
| | | (NH4)2SO4 → NH4HSO4 + NH3 (Δm = −12.9%) | (7) |
| | | NH4HSO4 → NH3SO3 + H2O (Δm = −15.7%) | (8) |
where SA is derived from dehydration of ABS (
eqn. (8)). This was confirmed in separate experiments in which ABS was held at 300 °C in dry and humid air (3 vol% H
2O), as shown in
Fig. 3. While the weight loss in dry air was consistent with SA formation, the presence of a low level of moisture restricted the loss to just 8% in 1 h, hinting at equilibrium control of dehydration. FTIR confirmed that only water was evolved. The most likely possibility is the formation of ammonium pyrosulfate:
39| | | 2NH4HSO4 → (NH4)2S2O7 + H2O (Δm = −7.8%) | (9) |
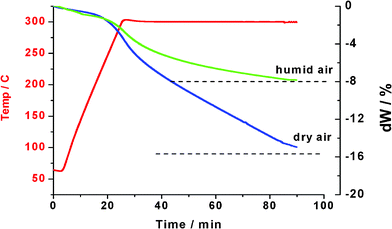 |
| | Fig. 3 TG-FTIR: ammonium bisulfate in dry and humid air at 300 °C. | |
Based on these studies, the thermal activation and degradation of ammonium sulfate, and the influence of humidity in stabilizing the active flux components, is summarized conveniently in Fig. S3 (ESI†).
3.1b Flux reactions with serpentine and MgO.
TG-FTIR studies of representative extraction mixtures showed very similar trends to the foregoing results for the pure flux. The effect of heating up to 350, 400 and 450 °C (1 h hold) in separate experiments is shown in Fig. 4, where the weight scale is given relative to AS as a more sensitive marker.¶ In all cases, the onset of weight loss was at ∼300 °C and NH3 was evolved, indicative of AS → ABS conversion. At 350 °C in humid air (blue curve), the weight loss approached a limit of ∼35%, well short of the theoretical value of ∼50% (dashed line). At higher temperatures in dry air, a distinct break in the weight curves was evident around theoretical, but this was soon exceeded at 450 °C. In this case, the evolved gas (FTIR) analysis is also shown (inset). Curiously, an initial burst of N2O was not accompanied by SO2, which evolved in a complex manner slightly later. Assuming that it is created simultaneously with N2O (eqn. (6)), it appears that SO2 is selectively retained in the melt and that sulfation occurs partially as a gas–solid process.
![TG curves for lizardite–AS held between 300 and 450 °C [inset: FTIR response for dry air held at 450 °C (red TG curve)].](/image/article/2012/RA/c2ra01347a/c2ra01347a-f4.gif) |
| | Fig. 4 TG curves for lizardite–AS held between 300 and 450 °C [inset: FTIR response for dry air held at 450 °C (red TG curve)]. | |
To confirm that ABS is a key ingredient in the flux, control experiments were made with a MgO–ABS mix (1:1 mol:mol) at 400 °C in dry and humid air. The results are presented in Fig. 5. For MgO, the sulfation reaction was probably complete during the heat-up stage. Significant weight loss had occurred already by 200 °C, where little or no SA should have been generated according to the data in Fig. 3. Hence, it was taken as evidence for the onset of sulfation. Both curves show a clear discontinuity at the onset of the 400 °C hold, where the weight loss (30–35%) corresponded to full sulfation according to:
| | | MgO + NH4HSO4 → MgSO4 + NH3 + H2O (Δm = −30.4%) | (10) |
 |
| | Fig. 5 TG curves for MgO–ABS heated to 400 °C (dry/humid air). | |
Just as in the lizardite–AS case, weight loss continued during the extended hold at 400 °C. However, this was decidedly more pronounced in dry as compared to humid conditions. As a supporting indicator of the stabilizing effect of just 3 vol% water vapour, the amount of evolved SO2 was reduced by a factor of ∼4.
3.2 Gram-scale flux extraction
While gravimetry remained useful as a rough indicator of reaction degree, gram-scale studies were made primarily to obtain reliable estimates of extraction efficiency (by analysis of dissolved Mg), flux loss (by analysis of dissolved S), and to gain insight into the solid-state chemistry by XRD.
3.2.1 Extraction as epsomite (MgSO4·7H2O).
Extraction as epsomite according to eqn. (11) below:| | | Mg3Si2O5(OH)4 + 3(NH4)2SO4 → 3MgSO4 + 2SiO2 + 5H2O + 6NH3 | (11) |
predicts a theoretical weight loss of 28.5%.
As flux extraction involves a solid mineral dispersed in a molten salt, there is some risk of “settling-out” so the effect of spatial distribution was also assessed. Mixtures (1 g total) were either shaped into a heap in the central quarter of the alumina boat or spread evenly to cover the entire base. After reaction at 400 °C, the weight loss was slightly under theoretical at 24%. XRD (see Fig S4, ESI†) showed the product to consist mainly of epsomite [MgSO4·7H2O]. Minor peaks of hexahydrite [MgSO4·6H2O] and efremovite were also evident. The last is a double or Tutton salt, of formula (NH4)2Mg2(SO4)3. Little or no evidence was seen for unreacted AS or lizardite, whose reference patterns are also available in ESI (Fig. S4†). At 450 °C, the weight loss increased dramatically to 39%, indicating substantial loss of the flux. This observation was consistent with micro-scale testing by TG-FTIR (red curve in Fig. 4). XRD showed the epsomite to be almost pure now, evidently growing at the expense of efremovite (see Fig. 6).
 |
| | Fig. 6 XRD of product ex lizardite–AS (1:3) after 1 h at 450 °C. | |
It is assumed that the heptahydrate was formed from anhydrous MgSO4 upon ambient exposure. The latter is known to be a strong dehydrating agent.
For the control study, a stoichiometric mixture of MgO and AS was prepared as below:
| | | 3MgO + 3(NH4)2SO4 → 3MgSO4 + 6NH3 + 3H2O | (12) |
for which a weight loss of 30.2% is expected. The actual losses after 1 h at 400 °C (heaped and spread) and 450 °C (spread) were close to theoretical,
viz., 29.2, 32.9 and 34.3%, respectively. XRD showed that the product was epsomite, as observed for the mineral.
Results of ICP-OES analysis of aqueous extracts from the lizardite and the MgO control are summarized in Table 1. Since all relevant sulfates are soluble, the S analysis measures recovery of the product along with any residual extractant. Thus, the soluble Mg level would be expected to approach, but never exceed, the S level. Table 1 reveals that the limiting factor in Mg extraction is the recovery of the sulfate, which barely exceeded 60%. Furthermore, there was no clear trend in values with reaction temperature, implying that flux loss may already be significant at 400 °C. Thus, the recycling efficiency of AS under these conditions would be seriously impaired. As shown in the footnote to Table 1, the clearest evidence for settling out of the flux was seen in the wide range of S recovery at 400 °C between top and bottom sections of the heaped lizardite sample. This suggests future recourse to a rotary kiln to maintain a more homogeneous suspension in the melt. Results for the MgO control were generally better, with yields of both S and Mg approaching 70%. This would be expected as the flux had direct access to the oxide in its intrinsic form, as indicated already by TG-FTIR (see Fig. 5).
Table 1 Soluble Mg and S recovery by ICP-OES from lizardite–AS (1:3) and MgO–AS (1:1) controls treated at various extraction temperatures
| Mg source |
ICP-OES |
400 °C |
450 °C |
500 °C |
| Spread Heaped |
Spread Heaped |
Spread Heaped |
|
Mean of 55.0 and 61.7% for top and bottom section, respectively.
|
| Liz. |
%Mg |
48.1 |
58.4 |
53.1 |
48.2 |
60.3 |
55.9 |
| %S |
60.6 |
58.5a |
52.3 |
47.9 |
59.5 |
54.7 |
| MgO |
%Mg |
66.4 |
64.7 |
68.9 |
— |
— |
— |
| %S |
68.0 |
69.9 |
71.9 |
— |
— |
— |
3.2.2 Extraction as efremovite [(NH4)2Mg2(SO4)3].
In view of the previous evidence for its transient formation, efremovite was deliberately targeted as product by working at lower temperature in mixtures suitably enriched in AS. For lizardite and the MgO control, the relevant equations are:| | | Mg3Si2O5(OH)4 + 4.5(NH4)2SO4 → 1.5(NH4)2Mg2(SO4)3 + 2SiO2 + 6NH3 + 5H2O | (13) |
| | | 2MgO + 3(NH4)2SO4 → (NH4)2Mg2(SO4)3 + 4NH3 + 2H2O | (14) |
Theoretical weight losses are closely similar at 22.0% and 21.8%, respectively. Being a soluble double salt, efremovite is a potentially valuable alternative product although the requirement of higher proportions of extractant for full Mg yield imposes an extra burden on the materials cycle. Gravimetry showed a clear trend in sample recovery after treatment at progressively higher temperatures. For lizardite, the weight loss increased systematically from 15% at 360 °C up to 22% at 390 °C, the last being close to theoretical. However, a sudden increase to 38% occurred at 400 °C, marking the onset of flux loss at a significant rate. In contrast, the weight loss for the MgO mixture was close to theoretical even at 400 °C (23.0%), suggesting that sulfation competes favourably with flux loss in this case. The trend in elemental recovery was consistent with the foregoing indicators from gravimetry. The corresponding ICP-OES analyses are depicted graphically in Fig. 7. For lizardite, the S(ulfate) level remained over 70% from 360–390 °C, implying better control of extractant losses, but the sudden drop to 55% observed at 400 °C was anticipated. Mg recovery was initially modest but progressed to ∼70% by 390 °C, before falling along with the S loss at 400 °C. For the MgO control, the encouraging indications from gravimetry were confirmed, with Mg and S recoveries around 70% at 350 °C, rising to over 90% by 400 °C.
![ICP-OES soluble Mg and S from serpentine–AS (1 : 4.5) and MgO–AS (2 : 3) mixtures [stoichiometry for efremovite].](/image/article/2012/RA/c2ra01347a/c2ra01347a-f7.gif) |
| | Fig. 7 ICP-OES soluble Mg and S from serpentine–AS (1:4.5) and MgO–AS (2:3) mixtures [stoichiometry for efremovite]. | |
XRD revealed an interesting trend in the associated solid-state chemistry. As shown in Fig. 8, a structure identified as the double salt boussingaultite [(NH4)2Mg(SO4)2·6H2O: #01-074-0276] was dominant at 360 °C along with weak reflections due to unreacted AS and lizardite. At 370 °C, reflections due to efremovite first appeared and these progressively replace those of boussingaultite up to 420 °C (see Fig. 9). A vestigial level of lizardite remained but new peaks grew in at 2θ = 10.75° (d = 8.25 Å), 21.5° (4.12 Å) and 27.0° (3.30 Å), characteristic of an unusual form of SiO2 as co-product [#43-0745].40 However, unlike for the stoichiometric (1:3) mixture, no epsomite was produced in this case. Diffractograms for intermediate temperatures 370, 380 and 400 °C are available in the ESI† (Fig S5). A scheme itemizing the conditions and likely routes by which these double salts are progressively converted to magnesium sulfate is also available in the ESI† (Fig. S6).
![XRD of product ex lizardite–AS after 1 h at 360 °C [B = boussingaultite (blue), AS = ammonium sulfate (brown), L = lizardite (green)].](/image/article/2012/RA/c2ra01347a/c2ra01347a-f8.gif) |
| | Fig. 8 XRD of product ex lizardite–AS after 1 h at 360 °C [B = boussingaultite (blue), AS = ammonium sulfate (brown), L = lizardite (green)]. | |
![XRD of product ex lizardite–AS after 1 h at 420 °C [E = efremovite (red), B = boussingaultite (blue), L = lizardite (green)].](/image/article/2012/RA/c2ra01347a/c2ra01347a-f9.gif) |
| | Fig. 9 XRD of product ex lizardite–AS after 1 h at 420 °C [E = efremovite (red), B = boussingaultite (blue), L = lizardite (green)]. | |
The most emphatic message to be taken from these studies is the evident competition between (desired) sulfation and loss of extractant even at 400 °C. Whereas recovery as efremovite at this temperature from MgO is almost quantitative, the more complex pre-dissolution and extraction of reactive Mg (as oxide or Mg2+ ion) from the silicate matrix is evidently more intractable. Future experiments in the tubular furnace should fully evaluate the stabilizing effect of humidity, as observed here by TG-FTIR, and explore longer heat treatment.
Conclusions
By virtue of their rich chemistry with magnesium, ammonium salts are likely to become ubiquitous in the field of CO2 mineralization. The sulfate is particularly suited to silicate mineral activation, readily decomposing above 300 °C to the more chemically aggressive bisulfate (and/or pyrosulfate) as key components in the flux. A combination of TG-FTIR, XRD and ICP-OES has revealed much of the condensed-phase chemistry underlying the extraction process. The reaction proceeds at a workable rate above 350 °C via the intermediacy of soluble double salts progressively rich in Mg. As currently practised, the elevated temperatures necessary to recover MgSO4 directly (400–450 °C) result in only modest yields (<70%) due primarily to settling out effects in the melt and irreversible loss of the flux by decomposition and/or sublimation. Extraction as efremovite [(NH4)2Mg2(SO4)3] offers a promising alternative as it proceeds below 400 °C where flux loss is restricted. The beneficial effect of humidity in stabilizing the flux has been reported but requires more intensive investigation.
Acknowledgements
J. H. and R. Z. are grateful for a joint A*Star/Tekes operating grant [FI-10-014 - NEACAP project 2010-2013] under which this work was conducted. J. F. is grateful to the Harry Elvings legacy to enable a four-month research secondment at ICES Singapore in 2011.
References
-
K. S. Lackner, Comparative impacts of fossil fuels and alternative energy sources, in Issues in environmental science & technology, ed. R. E. Hester and R. M. Harrison, 2010, vol. 29, Carbon capture: sequestration & storage, RSC, Cambridge, UK Search PubMed.
- S. Stucki, A. Schuler and M. Constantinescu, Int. J. Hydrogen Energy, 1995, 20, 653 CrossRef CAS.
- K. S. Lackner, Washing Carbon out of the Air, Sci. Am., 2010, 302, 66 CrossRef CAS.
- M. Mahmoudkhani and D. W. Keith, Int. J. Greenhouse Gas Control, 2009, 3, 376 CrossRef CAS.
- K. S. Lackner, Science, 2003, 300, 1677 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 CAS.
- D. J. Allen and G. F. Brent, Environ. Sci. Technol., 2010, 44, 2735 CrossRef CAS.
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce Jr. and D. H. Sharp, Energy, 1995, 20, 1153 CrossRef CAS.
- K. S. Lackner, D. P. Butt and C. H. Wendt, Energy Convers. Manage., 1997, 38, S259 CrossRef CAS.
- S. J. Gerdemann, W. K. O'Connor, D. C. Dahlin, L. R. Penner and H. Rush, Environ. Sci. Technol., 2007, 41, 2587 CrossRef CAS.
- A.-H. Park, A. R. Jadhav and L.-S. Fan, Can. J. Chem. Eng., 2008, 81, 885 CrossRef.
- S. Kodama, T. Nishimoto, N. Yamamoto, K. Yogo and K. Yamada, Energy, 2008, 33, 776 CrossRef.
- H. Béarat, M. J. McKelvy, A. V. G. Chizmeshya, D. Gormley, R. Nunez, R. W. Carpenter, K. Squires and G. H. Wolf, Environ. Sci. Technol., 2006, 40, 4802 CrossRef.
- M. Kakizawa, A. Yamasaki and Y. Yanagisawa, Energy, 2001, 26, 341 CrossRef CAS.
- A.-H. Park and L. S. Fan, Chem. Eng. Sci., 2004, 59, 5241 CrossRef CAS.
- M. M. Maroto-Valer, D. J. Fauth, M. E. Kuchta, Y. Zhang and J. M. Andrésen, Fuel Process. Technol., 2005, 86, 1627 CrossRef CAS.
- M. Hänchen, V. Prigiobbe, G. Storti, T. M. Seward and M. Mazzotti, Geochim. Cosmochim. Acta, 2006, 70, 4403 CrossRef.
- S. C. M. Krevor and K. S. Lackner, Energy Proc., 2009, 1, 4867 CrossRef CAS.
- V. Prigiobbe, M. Hänchen, M. Werner, R. Baciocchi and M. Mazzotti, Energy Proc., 2009, 1, 4885 CrossRef.
- S. C. M. Krevor and K. S. Lackner, Int. J. Greenhouse Gas Control, 2011, 5, 1073 CrossRef CAS.
- V. Prigiobbe and M. Mazzotti, Chem. Eng. Sci., 2011, 66, 6544 CrossRef CAS.
- R. Zevenhoven, J. Fagerlund and J. K. Songok, Greenhouse Gases: Sci. Technol., 2011, 1, 48 CrossRef CAS.
-
E. Nduagu. MSc (Eng) thesis. Åbo Akademi University. Turku, Finland. 2008.
- E. Nduagu, T. Björklöf, J. Fagerlund, J. Wärnå, H. Geerlings and R. Zevenhoven, Miner. Eng., 2012, 30, 75 CrossRef CAS.
- E. Nduagu, T. Björklöf, J. Fagerlund, E. Mäkelä, J. Salonen, H. Geerlings and R. Zevenhoven, Miner. Eng., 2012, 30, 87 CrossRef CAS.
- R. Zevenhoven, S. Teir and S. Eloneva, Energy, 2008, 33, 362 CrossRef CAS.
- See, e.g., Magnesium compounds, in Kirk–Othmer Encyclopeadia of Chemical Technology, 2004, vol. 15, Wiley On-line Search PubMed.
- A. Botha and C. A. Strydom, Hydrometallurgy, 2001, 62, 175 CrossRef CAS.
-
P. Bai, J. Bu, T. Z. Yeo, P. Sharratt, A. Borgna and H. H. Khoo, ICCDU-XI, 2011, Dijon, France, June 27–30 Search PubMed.
- H. H. Khoo, P. N. Sharratt, J. Bu, Y. Yeo, A. Borgna, J. G. Highfield, T. Björklöf and R. Zevenhoven, Ind. Eng. Chem. Res., 2011, 50, 11350 CrossRef CAS.
- J. Meima, R. Vanderweijden, T. Eighmy and R. Comans, Appl. Geochem., 2002, 17, 1503 CrossRef CAS.
- T. Shahwan, B. Zűnbűl, A. E. Eroğlu and S. Yilmaz, Appl. Clay Sci., 2005, 30, 209 CrossRef CAS.
-
R. Zevenhoven, T. Björklöf, J. Fagerlund, I. Romão, J. Highfield and J. Bu, Proceedings of ACEME10, 2010, Turku, Finland, Nov. 29–Dec. 1 Search PubMed.
-
T. Björklöf. MSc (Eng) thesis. Åbo Akademi University. Turku, Finland. 2010.
- J. Fagerlund, J. Highfield and R. Zevenhoven, ChemSusChem Search PubMed , submitted.
-
J. Highfield, J. Bu, J. Fagerlund and R. Zevenhoven, 11th International Conference on Carbon Dioxide Utilization (ICCDU XI), Dijon, France, 27–30th June 2011, Poster 025 Search PubMed.
-
J. Highfield, H. Q. Lim, J. Fagerlund and R. Zevenhoven, RSC Adv., 2012 10.1039/C2RA20575K.
-
K. Yoshikubo, Sulfamic acid & sulfamates, in Kirk–Othmer encyclopeadia of chemical technology, Wiley On-line, 2001 Search PubMed.
- M. Jariwala, J. Crawford and D. J. LeCaptain, Ind. Eng. Chem. Res., 2007, 46, 4900 CrossRef CAS.
- S. Andrews, M. Papiz, R. McMeeking, A. Blake, B. Lowe, K. Franklin, J. Helliwell and M. Harding, J. Chem. Soc., Dalton Trans., 1988, 2513 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01347a |
| ‡ A Glossary of mineral names, chemical formulae, XRD reference (ICDD) codes, etc., is available in ESI† (SI Table S1). |
| § Abbreviations may be found in the Glossary.† |
| ¶ A mixture of lizardite–AS (1:3 mol:mol) reacting according to eqn. (1) has a theoretical weight loss of 28.5%, or 48.4% with respect to AS. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.