Straightforward, metal-free, and stereoselective synthesis of 9-oxo- and 10-hydroxy-2(E)-decenoic acids, important components of honeybee (Apis mellifera) secretions†
Ciro
Milite
a,
Monica
Viviano
a,
Marisabella
Santoriello
a,
Fabio
Aricò
b,
Gianluca
Sbardella
a and
Sabrina
Castellano
*a
aDipartimento di Scienze Farmaceutiche e Biomediche, Epigenetic Med Chem Lab, Università degli Studi di Salerno, Via Ponte Don Melillo, I-84084 Fisciano (SA), Italy. E-mail: scastellano@unisa.it; Fax: +39-089-96-9602; Tel: +39-089-96-9244
bDipartimento di Scienze Ambientali, Informatica e Statistica, Università Ca Foscari Venezia, 2137 Dorsoduro 30123 Venezia, Italy
First published on 13th March 2012
Abstract
10-Hydroxy-2E-decenoic (10-HDA) and 9-oxo-2E-decenoic (9-ODA) acids, two components identified in honeybee secretions, have both received considerable recent interest due to their involvement in caste switch and maintenance. Herein we report for the first time a metal-free, gram scale, and stereoselective synthesis of these honeybee secretion components by TEMPO catalyzed oxidation of readily available alcohols and subsequent Doebner–Knoevenagel reactions between the resulting aldehydes and malonic acid. Mechanistic investigations undertaken highlighted the crucial role of the Doebner–Knoevenagel reaction in the high yielding and selective preparation of the α,β-unsaturated acids 10-HDA and 9-ODA. The combination of inexpensive and environmentally friendly reagents with simple synthetic procedures renders this approach a valuable green strategy for the gram scale preparation of these biologically relevant natural molecules.
Introduction
Social insects like honeybees (Apis mellifera) have fascinated researchers for many years, particularly for their hierarchical structures. Secretions like royal jelly and the so-called “queen substance” are crucial for caste switch and maintenance. In fact, only queen larvae are fed exclusively on royal jelly and develop into sexually mature females, unlike the worker bees. Furthermore, the queen substance, produced by the queen, inhibits ovarian development in worker bees.1,2 Two major components identified, respectively, in royal jelly and queen substance are 10-hydroxy-2E-decenoic (1, 10-HDA) and 9-oxo-2E-decenoic (2, 9-ODA) acids (Fig. 1). These biologically active compounds are isolated from bees but in quantities not adequate for practical purposes and by employing complex purification procedures.3 As a result, in recent years, several synthetic approaches for 10-HDA and 9-ODA have been reported.4–8 However, in general these methodologies employ expensive and toxic reagents, or require procedures that are difficult to handle on a large scale.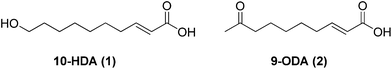 | ||
| Fig. 1 Structures of 10-hydroxy-2E-decenoic acid (10-HDA, 1) and 9-oxo-2E-decenoic acid (9-ODA, 2). | ||
In this work, we report for the first time a cost-effective and metal-free strategy for the selective preparation of compounds 1 and 2 on a multigram scale. An efficient synthetic route to these molecules will help clarify their role in the phenotypical shift from worker to queen bees.9
Results and discussion
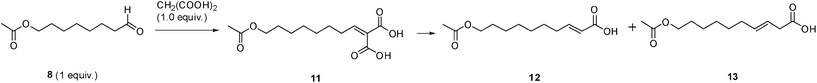
We based our approach to the synthesis of the α,β-unsaturated acids 1 and 2 on the common retrosynthetic pathway involving the decarboxylative Knoevenagel condensation (Doebner–Knoevenagel reaction) between malonic acid and 8-hydroxyoctanal 3 (for compound 1) or 7-oxooctanal 4 (for compound 2). The aldehydes 3 and 4 could be obtained from the oxidation of the corresponding alcohols octane-1,8-diol 5 and 7-oxooctan-1-ol 6, respectively (Scheme 1).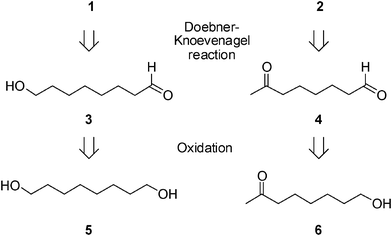 | ||
| Scheme 1 Retrosynthetic analysis of 10-HDA (1) and 9-ODA (2). | ||
However, as the Doebner–Knoevenagel reaction of hydroxyaldehydes is generally low yielding, we opted to synthesize compound 1 starting from the hydroxyl-protected aldehyde 8. This was prepared in two steps (Scheme 2): selective protection of the commercially available octane-1,8-diol 5 followed by oxidation of the monoprotected diol 7.10 The choice of the proper oxidation procedure was a critical step for the success of our strategy as we were aiming at a highly efficient and metal-free synthetic method. Aldehyde 8 could be obtained in quantitative yield by oxidation with 1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one (Dess–Martin periodinane, DMP), a green alternative to metal-based oxidizing agents.11 However, besides being an expensive reagent, DMP is potentially explosive12 and therefore not suitable at a multigram scale. On the other hand, among the various oxidants for alcohols, oxidation with 2,2,6,6-tetramethylpiperidine-N-oxy (TEMPO) is very appealing since it is a non-metallic catalyst and, foremost, it is environmentally friendly and economical.13 Indeed, when the oxidation of the monoprotected diol 7 was performed employing TEMPO with a stoichiometric amount of bleach (NaClO) as terminal oxidant, the hydroxyl-protected aldehyde 8 was selectively formed in quantitative yield (Scheme 2).
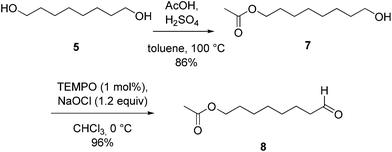 | ||
| Scheme 2 Preparation of aldehyde 8. | ||
At the same time, the preparation of aldehyde 4, required for the synthesis of compound 2, was straightforwardly obtained in three steps (Scheme 3). In particular the starting 5-bromopentan-1-ol (9) was coupled with 3-oxobutanoate to achieve the β-ketoester 10 which, after saponification followed by acid catalyzed decarboxylation, gave the alcohol 6. Finally, TEMPO oxidation furnished the desired 7-oxooctanal 4.
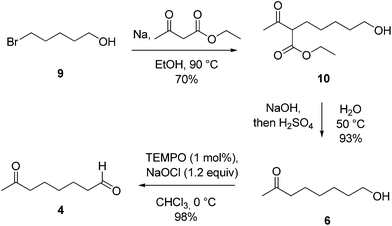 | ||
| Scheme 3 Preparation of aldehyde 4. | ||
Having successfully synthesized aldehydes 4 and 8, we next focused on the Doebner–Knoevenagel reaction. This synthetic approach to α,β-unsaturated acids presents some important drawbacks:14 the yields for enolizable aldehydes are usually low, the reaction conditions generally require a large excess of malonic acid and elevated temperatures, and the E/Z-selectivity varies. Furthermore, the major disadvantage is the production of mixtures of α,β- and β,γ-unsaturated derivatives. On the other hand, the Doebner–Knoevenagel reaction offers important advantages over other alternatives, i.e., the use of inexpensive reagents and a high atom economy. Moreover, it can be used on a large scale. Indeed, the Doebner–Knoevenagel reaction has been widely reported for the synthesis of 10-HDA.5,7,8 However, the literature is quite ambiguous. To the best of our knowledge, there are no definite remarks about chemo- and stereoselectivity and quite often the results are not supported by a detailed experimental section. In addition, the mechanistic aspects of the reaction are still controversial. Therefore, we set up a series of experiments to identify the best conditions, in terms of efficiency and selectivity, for the reaction between aldehyde 8 and malonic acid to give the desired α,β-unsaturated acid 12, a protected derivative of target compound 1. The results obtained are summarized in Table 1.
| Entry | Solvent/catalyst | Co-catalyst | T (°C) | Time (h) | 11 yield (%)a | 12 + 13 yield (%)a | Ratio 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13b :13b |
|---|---|---|---|---|---|---|---|
| a Chromatographically isolated yield relative to aldehyde 8. b Detected by 1H NMR. c Conventional heating. d Microwave irradiation. | |||||||
| 1 | Pyridine (solvent) | Piperidine (1 equiv.) | Reflux | 3 | 10 | 68 | 62:38 |
| 2 | Pyridine (solvent) | Piperidine (1 equiv.) | r.t. | 72 | 21 | 66 | 64:36 |
| 3 | Pyridine (1.3 equiv.) | Piperidine (0.01 equiv.) | Reflux | 3 | — | 88 | 86:14 |
| 4 | Pyridine (1.3 equiv.) | Piperidine (0.01 equiv.) | r.t | 72 | — | 92 | 95:5 |
| 5 | DMF | DMAP | r.t. | 72 | — | 82 | ≥1:99 |
| 6 | DMF | DMAP | 80 °Cc | 3 | 52 | 16 | 50:50 |
| 7 | DMF | DMAP | 80 °Cd | 3 × 0.5 | 15 | 60 | 45:55 |
| 8 | — | — | r.t. | 24 | 4 | — | — |
| 9 | — | Piperidine (0.01 equiv.) | r.t. | 24 | 50 | — | — |
| 10 | Pyridine (1.3 equiv.) | — | r.t. | 72 | — | 89 | ≥99:1 |
| 11 | Pyridine (1.3 equiv.) | — | 40 °Cd | 3 × 0.5 | — | 53 | ≥99:1 |
| 12 | Pyridine (1.3 equiv.) | — | Reflux | 3 | — | 20 | 83:17 |
| 13 | Pyridine (1.3 equiv.) | — | 80 °Cd | 3 × 0.5 | — | 41 | 75:25 |
Standard Doebner–Knoevenagel conditions, employing pyridine used as a solvent in the presence of piperidine (1.0 equiv.) at reflux temperature, gave excellent stereoselectivity (E/Z ≥ 99:1) but low chemoselectivity (α,β:β,γ = 62:38) and moderate yields, due to the incomplete decarboxylation of alkylidenemalonic acid 1115 (Table 1, entry 1). Lowering temperature and increasing reaction time (Table 1, entry 2) did not provide better results. When a reduced amount of pyridine (1.3 equiv.) and catalytic piperidine (0.01 equiv.) were used, the yield was almost quantitative and selectivity increased, yet to a level not adequate for a pharmaceutical grade compound (Table 1, entries 3–4). The replacement of piperidine with dimethylaminopyridine (DMAP), which has been described as an excellent catalyst for the Doebner–Knoevenagel synthesis of α,β-unsaturated esters,14 did not give the desired results (Table 1, entries 5–7). In fact, at room temperature, both yield and selectivity were excellent, although the β,γ-unsaturated acid 1316 was obtained instead of the α,β-unsaturated acid. The desired α,β-unsaturated acid 12 could be obtained by performing the reaction at a higher temperature, but in low yield and, above all, poor selectivity. Using microwave irradiation instead of conventional heating actually improved the yield, but not the α,β:β,γ ratio. These results prompted us to reconsider the possible mechanisms for the reaction.
In the early 1950s, Corey17,18 demonstrated that alkylidenemalonic acid derivatives 14 undergo decarboxylation in pyridine at 110 °C by a three-step process involving isomerization to a β,γ-unsaturated malonic acid derivative 15, anionic decarboxylation and protonation of the resulting mesomeric anion to afford a mixture of α,β- and β,γ-unsaturated products 16 and 17 (Scheme 4). The rate-determining step in the conversion of 14 to the β,γ-isomer 15 is the abstraction of a proton from the un-ionized acid. It is likely that this mechanism operates during the reaction of malonic acid and aldehyde 8 at high temperature or in the presence of a strong base. More recently, Kemme et al.6 proposed an alternative two-step mechanism for the selective formation of the α,β-unsaturated acids by the Doebner–Knoevenagel reaction. These authors suggested that in the presence of a catalytic amount of pyrrolidine at room temperature a fast classical Knoevenagel condensation occurs, with formation of alkylidenemalonic acid derivatives 14. The second step is a slower decarboxylation, initiated by pyridine functioning as a Lewis basic catalyst, with the selective formation of α,β-unsaturated derivative 16 (Scheme 5).
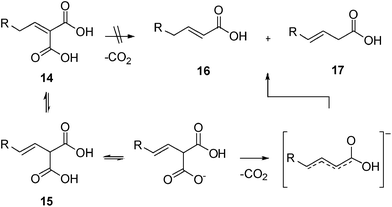 | ||
| Scheme 4 Decarboxylation of α,β-unsaturated malonic acids via β,γ- unsaturated intermediate. | ||
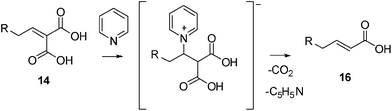 | ||
| Scheme 5 Decarboxylation of α,β-unsaturated malonic acids catalyzed by pyridine. | ||
We decided then to verify this latest reaction mechanism in order to improve the selective preparation of the desired product 12. Thus, first we carried out a reaction in the absence of pyridine (Table 1, entries 8, 9). Under neat reaction conditions (Table 1, entry 8), the formation of 11 is very slow (4% conversion after 24 h at room temperature), while with a catalytic amount of piperidine (Table 1, entry 9) the reaction is faster and no decarboxylation product was detected. Furthermore compound 11, upon heating in DMF, gave exclusively the β,γ-isomer 13 whereas reaction with pyridine at room temperature furnished selectively the α,β-unsaturated derivative 12.
According to the results obtained we decided to perform the reaction in the absence of piperidine using pyridine (1.3 equiv.) as the sole reagent in order to avoid the isomerization step and push toward the selective formation of α,β-unsaturated derivative 12. Remarkably, when the reaction was carried out at room temperature for 72 hours (Table 1, entry 10) the decarboxylation of alkylidenemalonic acid 11 was complete and acid 12 was obtained in good yield with high selectivity (α,β:β,γ ≥ 99:1). When we attempted to reduce the reaction time by microwave irradiation at 40 °C (Table 1, entry 11) the selective formation of the desired α,β-unsaturated acid 12 was still obtained but in a lower yield (starting aldehyde 8 was recovered). Consistently, increasing temperature, using both conventional heating and microwave irradiation (Table 1, entries 12, 13), was detrimental for the chemoselectivity. In addition, it is worth noting that also under these conditions (absence of piperidine) all the reactions were characterized by complete stereoselectivity (E/Z ≥ 99:1).
After optimization studies, α,β-unsaturated acids 1 and 2 were prepared on a multigram scale. In fact, after treating acid 12 with potassium carbonate in methanol at room temperature, deprotected target compound 1 was obtained in quantitative yield. α,β-Unsaturated acid 2 was obtained in excellent yield (87%) and selectivity (α,β:β,γ ≥ 99:1) from aldehyde 4 under our optimized Doebner–Knoevenagel conditions (see Experimental section for details).
Conclusions
In summary, herein we described an efficient and selective approach to the synthesis of 10-hydroxy-2E-decenoic (1, 10-HDA) and 9-oxo-2E-decenoic (2, 9-ODA) acids by the Doebner–Knoevenagel reaction of aldehydes with malonic acid. TEMPO-catalyzed oxidation allowed the synthesis of aldehydes in high yield using inexpensive and metal-free reagents. Optimized conditions for the Doebner–Knoevenagel reaction provided the two honeybee metabolites in high yield and excellent selectivity. The use of inexpensive and environmental friendly reagents, together with the simple synthetic procedures employed, make this procedure a valuable alternative for the preparation of 10-HDA and 9-ODA on a large scale.Experimental
General information
All chemicals were purchased from Sigma Aldrich (Milan, Italy). Reactions were routinely monitored by TLC performed on aluminum-backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254) using a KMnO4 alkaline solution. Solvents were removed using a rotary evaporator operating at a reduced pressure of ∼10 Torr. Organic solutions were dried over anhydrous Na2SO4. Chromatographic separations were performed on silica gel (silica gel 60, 0.015–0.040 mm; Merck DC) columns. Melting points were determined on a Stuart SMP30 melting point apparatus in open capillary tubes and are uncorrected. 1H NMR spectra were recorded at 300 MHz on a Bruker Avance 300 spectrometer. Chemical shifts are reported in δ (ppm) relative to the internal reference tetramethylsilane (TMS). Mass spectra were recorded on a Finnigan LCQ DECA TermoQuest (San Jose, USA) mass spectrometer in electrospray positive and negative ionization modes (ESI-MS). Purity of tested compounds was established by combustion analysis, performed on a Perkin-Elmer 2400 CHN elemental analyzer at the laboratory of microanalysis of the Department of Chemistry and Biology, University of Salerno (Italy); the analytical results were within ±0.4% of the theoretical values. When the elemental analysis is not included, compounds were used in the next step without further purification.8-Hydroxyoctyl acetate (7)
To a solution of 1,8-octanediol (10.00 g, 68.38 mmol) in toluene (200 mL), water (7.00 mL), AcOH (4.07 mL, 71.12 mmol) and H2SO4 (0.15 mL, 2.80 mmol) were added. The resulting mixture was stirred at reflux for 48 h. After cooling at room temperature, the organic layer was separated, washed with NaHCO3 (80 mL), water (80 mL) and brine (80 mL), dried (Na2SO4), filtered, and concentrated in vacuo. Purification by column chromatography on silica gel (DCM:EtOAc 95:5) provided the title compound 7 (11.08 g, 86%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.04 (t, J = 6.7 Hz, 2H), 3.63 (t, J = 6.6 Hz, 2H), 2.04 (s, 3H), 1.68–1.49 (m, 5H, one exchangeable with D2O), 1.43–1.25 (m, 8H). MS (ESI) m/z: 189 [M + H]+. Data are in good agreement with the literature.19
Ethyl 2-acetyl-7-hydroxyheptanoate (10)
To a solution of Na (1.37 g, 59.56 mmol) in dry ethanol (35 mL), ethyl 3-oxobutanoate (7.59 mL, 59.56 mmol) was added. The mixture was stirred at reflux for 1 h and 5-bromo-1-pentanol (7.21 mL, 59.56 mmol) was added. The resulting mixture was stirred at reflux for an additional 3 h and, after cooling at room temperature, the solvent was evaporated. The crude residue was dissolved in EtOAc (200 mL), filtered and concentrated in vacuum. Purification by column chromatography on silica gel (DCM:EtOAc 40:60) provided the title compound 10 (9.27 g, 72%) as a pale yellow oil. 1H NMR (300 MHz, CDCl3) δ 4.20 (q, J = 7.1 Hz, 2H), 3.71–3.57 (m, 2H), 3.40 (t, J = 7.1 Hz, 1H), 2.22 (s, 3H), 1.91–1.78 (m, 2H), 1.66–1.50 (m, 3H, one exchangeable with D2O), 1.44–1.17 (m, 7H). MS (ESI) m/z: 217 [M + H]+.
8-Hydroxyoctan-2-one (6)
A suspension of 2-acetyl-7-hydroxyeptanoate (10) (9.20 g, 42.54 mmol) in 0.5 M aqueous solution of NaOH (215 mL, 107.0 mmol) was stirred at room temperature. After the dissolution was completed, a 0.2 M aqueous solution of H2SO4 (400 mL) was added and the mixture was stirred at 50 °C for 30 min. After cooling at room temperature, the reaction was extracted with EtOAc (3 × 200 mL). The combined organic phases were washed with brine (100 mL), dried (Na2SO4) and filtered. Evaporation of the solvent gave the title compound 6 (5.70 g, 93%) as a pale yellow oil. 1H NMR (300 MHz, CDCl3) δ 3.70–3.58 (m, 2H), 2.43 (t, J = 7.3 Hz, 2H), 2.13 (s, 3H), 1.76–1.47 (m, 5H, one exchangeable with D2O), 1.44–1.22 (m, 4H). MS (ESI) m/z: 145 [M + H]+. Data are in good agreement with the literature.20General procedure for TEMPO-catalyzed oxidation of alcohols to aldehydes
To a solution of the proper alcohol (53.10 mmol) in chloroform (100 mL), TEMPO (0.53 mmol) and a 3% aqueous solution of NaOCl (157 mL, 66.37 mmol) were added. NaOCl solution pH was adjusted to 8.6 by dissolving 50 mg mL−1 of NaHCO3. The resulting mixture was stirred at 0 °C for 30 min. The organic phase was separated, washed with 10% aqueous solution of Na2S2O3 (3 × 50 mL) and brine (80 mL), dried (Na2SO4), filtered, and concentrated in vacuo. Purification by column chromatography on silica gel (DCM:AcOEt 90:10) provided the title compounds.
8-Acetoxyoctanal (8)
Colorless oil, 96% yield. 1H NMR (300 MHz, CDCl3) δ 9.74 (s, 1H, exchangeable with D2O), 4.02 (t, J = 6.7 Hz, 2H), 2.40 (td, J = 7.3, 1.5 Hz, 2H), 2.02 (s, 3H), 1.70–1.54 (m, 4H), 1.44–1.27 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 202.23, 170.76, 64.11, 43.48, 28.66, 28.61, 28.20, 25.37, 21.62, 20.61. MS (ESI) m/z: 187 [M + H]+. Data are in good agreement with the literature.217-Oxooctanal (4)
Colorless oil, 98% yield. 1H NMR (300 MHz, CDCl3) δ 9.75 (s, 1H, exchangeable with D2O), 2.47–2.40 (m, 4H), 2.12 (s, 3H), 1.70–1.52 (m, 4H), 1.40–1.26 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 209.26, 202.99, 44.27, 43.96, 30.49, 29.24, 24.05, 22.44. MS (ESI) m/z: 143 [M + H]+. Data are in good agreement with the literature.20Optimized procedure for Doebner–Knoevenagel reactions
Malonic acid (15.30 mmol) and pyridine (19.89 mmol) were added to aldehyde (15.30 mmol) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 72 h. The resulting mixture was taken up with water (15 mL), acidified to pH 1 with 2 N aqueous solution of HCl and extracted with AcOEt (3 × 40 mL). The combined organic phases were washed with brine (100 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification by column chromatography on silica gel (DCM:AcOEt 50:50) provided the title compounds.
(E)-10-Acetoxydec-2-enoic acid (12)
Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.07 (dt, J = 15.6, 6.9 Hz, 1H), 5.82 (d, J = 15.6 Hz, 1H), 4.05 (t, J = 6.6 Hz, 2H), 2.27–2.18 (m, 2H), 2.05 (s, 3H), 1.67–1.56 (m, 2H), 1.52–1.42 (m, 2H), 1.39–1.29 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 171.75, 171.34, 152.13, 120.84, 64.61, 32.28, 29.03, 28.63, 27.85, 25.85, 21.02. MS (ESI) m/z: 229 [M + H]+.(E)-9-Oxodec-2-enoic acid (2)
White solid (Et2O/cyclohexane), mp: 49–51 °C. 1H NMR (300 MHz, CDCl3): δ 11.13 (br s, 1H, exchangeable with D2O), 7.06 (dt, J = 15.3, 6.9 Hz, 1H), 5.82 (d, J = 15.6 Hz, 1H), 2.43 (t, J = 7.3 Hz, 2H), 2.30–2.19 (m, 2H), 2.14 (s, 3H), 1.65–1.53 (m, 2H), 1.52–1.42 (m, 2H), 1.39–1.23 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 209.85, 172.35, 152.38, 121.56, 44.10, 32.65, 30.44, 29.23, 28.29, 24.08. MS (ESI) m/z: 185 [M + H]+. Anal. Calcd for C10H16O3: C, 65.19; H, 8.75. Found: C, 65.36; H, 8.78. Data are in good agreement with the literature.6(E)-10-Hydroxydec-2-enoic acid (1)
To a solution of (E)-10-acetoxydec-2-enoic acid (12) (2.28 g, 10.00 mmol) in methanol (10 mL), K2CO3 (1.52 g, 11.00 mmol) was added. The mixture was stirred at room temperature for 12 h and then the solvent was evaporated. The crude material was taken up with water (10 mL), acidified to pH 1 with 2 N aqueous solution of HCl and extracted with AcOEt (3 × 10 mL). The combined organic phases were washed with brine (100 mL), dried (Na2SO4), filtered and concentrated in vacuo. Trituration with cyclohexane yielded a white solid (1.82 mg, 98%) which was recrystallized from Et2O/cyclohexane. M.p.: 64–65 °C. 1H NMR (300 MHz, CDCl3) δ 7.06 (dt, J = 15.6, 6.9 Hz, 1H), 6.47 (br s, 2H, exchangeable with D2O), 5.82 (d, J = 15.6 Hz, 1H), 3.64 (t, J = 6.6 Hz, 2H), 2.28–2.17 (m, 2H), 1.62–1.28 (m, 10H). 13C NMR (75 MHz, CDCl3) δ 171.23, 151.84, 120.97, 62.87, 32.57, 32.30, 29.19, 29.13, 27.91, 25.67. MS (ESI) m/z: 187 [M + H]+. Anal. Calcd for C10H18O3: C, 64.49; H, 9.74. Found: C, 64.58; H, 9.77. Data are in good agreement with the literature.22Acknowledgements
This work was supported by grants from Ministero dell'Università e della Ricerca Scientifica e Tecnologica – PRIN 2008 (S.C.), Università di Salerno (G.S., S.C.) and COST Action TD09/05 Epigenetics (G.S.).References
- R. Kucharski, J. Maleszka, S. Foret and R. Maleszka, Science, 2008, 319, 1827–1830 CrossRef CAS.
- Y. Y. Shi, Z. Y. Huang, Z. J. Zeng, Z. L. Wang, X. B. Wu and W. Y. Yan, PLoS One, 2011, 6, e18808 CAS.
- N. Noda, K. Umebayashi, T. Nakatani, K. Miyahara and K. Ishiyama, Lipids, 2005, 40, 833–838 CrossRef CAS.
- G. Ishmuratov, M. Yakovleva, K. Tambovtsev, Y. Legostaeva, L. Kravchenko, N. Ishmuratova and G. Tolstikov, Chem. Nat. Compd., 2008, 44, 74–76 CrossRef CAS.
- G. Y. Ishmuratov, R. Y. Kharisov, O. V. Botsman, N. M. Ishmuratova and G. A. Tolstikov, Chem. Nat. Compd., 2002, 38, 1–23 CrossRef CAS.
- S. T. Kemme, T. Šmejkal and B. Breit, Adv. Synth. Catal., 2008, 350, 989–994 CrossRef CAS.
- R. Y. Kharisov, O. V. Botsman, L. P. Botsman, N. M. Ishmuratova, G. Y. Ishmuratov and G. A. Tolstikov, Chem. Nat. Compd., 2002, 38, 145–148 CrossRef CAS.
- G. I. Fray, R. H. Jaeger, E. D. Morgan, R. Robinson and A. D. B. Sloan, Tetrahedron, 1961, 15, 18–25 CrossRef.
- A. Spannhoff, Y. K. Kim, N. J. M. Raynal, V. Gharibyan, M.-B. Su, Y.-Y. Zhou, J. Li, S. Castellano, G. Sbardella, J.-P. J. Issa and M. T. Bedford, EMBO Rep., 2011, 12, 238–243 CrossRef CAS.
- A. Wicki Markus, E. Nielsen Kent, Use of a biphasic solvent system in a selective process for preparing monoesters from diols, U.S. Pat. Appl. Publ., 20050143599, 30 June, 2005 Search PubMed.
- C. Milite, S. Castellano, R. Benedetti, A. Tosco, C. Ciliberti, C. Vicidomini, L. Boully, G. Franci, L. Altucci, A. Mai and G. Sbardella, Bioorg. Med. Chem., 2011, 19, 3690–3701 CrossRef CAS.
- J. B. H. Plumb, Chem. Eng. News, 1990, 68, 3 CAS.
- P. Lucio Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef CAS.
- B. List, A. Doehring, M. T. Hechavarria Fonseca, A. Job and R. Rios Torres, Tetrahedron, 2006, 62, 476–482 CrossRef CAS.
- 2-(8-Acetoxyoctylidene)malonic acid (11). Colorless oil, 1H NMR (300 MHz, CDCl3) δ 7.88 (t, J = 7.5 Hz, 1H), 6.47 (br s, 2H, exchangeble with D2O), 4.06 (t, J = 6.7 Hz, 2H), 2.90–2.78 (m, 2H), 2.06 (s, 3H), 1.74–1.51 (m, 6H), 1.47–1.22 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 171.10, 169.57, 168.20, 164.43, 122.65, 65.57, 31.48, 29.79, 29.43, 29.06, 28.85, 26.30, 21.57. MS (ESI) m/z: 273 [M + H]+.
- (E)-10-Acetoxydec-3-enoicacid (13). Colorless vitreous solid, 1H NMR (300 MHz, CDCl3) δ 9.31 (br s, 1H, exchange with D2O), 5.65–5.44 (m, 2H), 4.05 (t, J = 6.6 Hz, 2H), 3.07 (d, J = 5.9 Hz, 2H), 1.98 (s, 3H), 1.65–1.48 (m, 2H), 1.34–1.17 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 177.38, 171.45, 134.88, 121.18, 64.59, 37.69, 32.21, 28.83, 28.57, 28.43, 25.62, 20.82. MS (ESI) m/z: 229 [M + H]+.
- E. J. Corey, J. Am. Chem. Soc., 1952, 74, 5897–5905 CrossRef CAS.
- E. J. Corey, J. Am. Chem. Soc., 1953, 75, 1163–1167 CrossRef CAS.
- J. H. Babler and M. J. Coghlan, Tetrahedron Lett., 1979, 20, 1971–1974 CrossRef.
- J. S. Dickschat, E. Helmke and S. Schulz, Chem. Biodiversity, 2005, 2, 318–353 CAS.
- G. Huang, J. Li, J. Lu and H. Aisa, Chem. Nat. Compd., 2006, 42, 727–729 CrossRef CAS.
- S. Matsui, Bull. Chem. Soc. Jpn., 1984, 57, 426–434 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. Copies of 1H and 13C NMR spectra of synthesized compounds. See DOI: 10.1039/c2ra20275a |
| This journal is © The Royal Society of Chemistry 2012 |