Free-standing Ni-microfiber-supported carbon nanotube aerogel hybrid electrodes in 3D for high-performance supercapacitors†
Yuzhu
Fang
a,
Fangting
Jiang
a,
Hong
Liu
b,
Xiaoming
Wu
b and
Yong
Lu
*a
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, Shanghai 200062, P. R. China. E-mail: ylu@chem.ecnu.edu.cn; Fax: (+86) 21 62233424; Tel: (+86) 21 62233424
bTANTZ Environmental Protection Technologies Ltd., 189 South Dunhe Road, Guangzhou 510300, P. R. China
First published on 15th May 2012
Abstract
Macroscopic Ni-microfiber-supported carbon nanotube aerogels (CNAGs) have been developed and demonstrate great potential as supercapacitor electrodes. The macroscopic carbon nanotubes (CNTs) are controllably prepared by a catalytic chemical vapour deposition method through CNT growth on a sinter-locked microfibrous structure (SMF) consisting of 5 vol% 8 μm Ni fibers. Polyimide is coated onto the CNTs rooted on the SMF-Ni by an impregnation/polymerization method and is subsequently carbonized by pyrolysis to create self-supporting CNAGs/SMF-Ni composite electrodes, wherein the Ni-fiber network serves as current collector and the CNTs grown on the Ni fiber act as nano conducting wires to link the charge-storage carbon aerogel (CAG) particles. This novel approach permits desirable large-area fabrication and provides a unique combination of high CNAG-loading (up to 71.0 wt%; CAG/CNT: 1.44 wt/wt), binder-free feature, excellent electrical conductivity, large surface area, macro-/meso-/micro-sized hierarchical porous structure and high permeability. A typical hybrid consisting of 68.5 wt% CNAG (CAG/CNT: 1.17 wt/wt) delivers not only a good capacitance (e.g., 359 F per gram CAG) at high rates but also excellent long-cycle life (5% loss after 300 cyclic voltammogram cycles and then almost unchanged through 1000 cycles).
Introduction
Carbon materials are widely used as electrode materials of energy-storage devices such as supercapacitors and batteries.1 Supercapacitors have many advantages over batteries, including high power density, longer cycle-life, a very simple charging circuit, low-cost maintenance, and operational safety. Significant improvement of their capacitance for achieving high energy density, however, is an important demand and remains challenging.2–5 This provides particular impetus for research on new types of carbon materials.2–14It is well known that the performance of carbon-based supercapacitors is strongly dependent on the carbon properties such as surface area, pore size distribution and tortuosity of pores.15 Whereas activated carbon (AC) is widely used for supercapacitor electrodes, the fraction of ultramicropores, which are inaccessible to the electrolyte ions, is very high, significantly lowering the specific capacitance of AC-based supercapacitors. Carbon aerogels (CAGs) are novel ultralight materials that have gained increased interest in recent years as a very attractive alternative to activated carbon for many applications such as energy storage.6,16–18 This interest stems largely from their high surface area, continuous porosities with characteristic pore diameters between 3 and 25 nm, and high electrical conductivity.19,20 In general, CAGs with different microstructures are prepared by the pyrolysis of organic polymers such as phenolic resins,20,21 pitches22 and polyimide.23 Among them, polyimide is widely used for the synthesis of CAGs because its preparation is simple and the original shape can be preserved even though accompanied by significant shrinkage during the carbonization process. For example, Takeichi et al.23,24 reported the CAGs obtained from the pyrolysis of porous polyimide films at 900 °C and they found that the size of the pores could be controlled by adjusting the composition of the polyimide films.
Despite the above advances, the practical application of CAGs in future technologies is hampered somewhat by some natural shortcomings.18 A CAG network consisting of agglomerate particles linked by covalent bridges unfortunately induces a high internal resistance within the aerogels (being detrimental for electrochemistry applications), due to the existence of the contact boundary between the carbon particles. In addition, the integration of CAGs into a macroscopic structure to translate the phenomenon at the nanoscale to the macroscopic level is required in real world devices rather than powders. Indeed, identifying novel carbon-based composites7–9,14,17,18 which permit desirable large-area fabrication and provide a unique combination of excellent electrical conductivity (to reduce ohmic losses and heating), large surface area accessible to electrolyte ions and macro-/meso-sized hierarchical porous structure favourable for ion transfer has been largely overlooked by the electrochemical energy-storage research community.
To accomplish this goal, some efforts have been devoted to the synthesis of carbon nanotube aerogels (CNAGs) through CNT-adding polymer pyrolysis, wherein the CNTs serve as not only nanocollectors for charge but also as network reinforcement accessories for achieving a robust macroscopic structure.17,18,25 Béguin et al.17 reported a CNAG monolithic structure obtained by one-step pyrolysis of pellets formed by pressing CNT–polyacrylonitrile blends, demonstrating interesting pseudo-capacitance properties but a low specific capacitance of 100 F g−1 in 1 mol L−1 H2SO4. Bordjiba et al.25 reported the synthesis of CNAGs by carbonizing a suspension of CNTs in dimethylformamide and demonstrated a high capacitance of 218 F g−1 for an optimum composite containing 3 wt% CNTs. However, such dispersion–carbonization preparation processes remain significantly challenging as yet, especially in obtaining a unique dispersion of CNTs in the aerogel matrix without destroying their integrity or reducing their aspect ratio.18 Bordjiba et al.18 also reported a new class of macroscopic CNAG structure obtained by coating CNTs/carbon-paper with carbon aerogels. Although such CNAGs/carbon-paper delivered a very high specific capacitance of 524 F g−1 in 1 mol L−1 H2SO4 that is almost four times that of CAGs (134 F g−1), its capacitance on the basis of the overall composite weight is very small (much smaller on the basis of the overall volume), due to the low CNAGs loading amount on the carbon fiber matrix.18,26 This would significantly increase the overall weight and volume of the energy-storage devices. Nevertheless, as a strategy, this method is successful and effective for integrating nanostructured materials (e.g., CAGs, CNTs and metal oxides) into macroscopic monoliths, bringing high-performance electrochemical energy-storage devices a step closer to reality.7–9,14,17,18
Herein, we demonstrate a new large-area CNAG embedded on sinter-locked 8 μm Ni fibers (SMF-Ni) for supercapacitors on a macroscopic scale obtained by a simple and cheap means as illustrated in Scheme 1. We obtain a CNTs/SMF-Ni composite with CNT loading of 50 wt% first through direct growth of CNTs on the SMF-Ni matrix via a catalytic chemical vapour deposition (CCVD) method using ethylene as the carbonaceous source.27 Afterwards, the CAG with loading as high as ∼42 wt% (corresponding to CNAG loading of 71 wt%) is assembled onto the CNTs rooted on the SMF-Ni matrix by polyimide-sol coating/pyrolysis processes (Table 1). This approach permits the desired macroscopic fabrication of self-supporting CNAG-based electrodes in a simple and cost-effective way and provides a unique combination of binderlessness, excellent conductivity, hierarchical macro-/mesoporous structure with a large mesopore surface area. Our CNAGs/SMF-Ni composites as supercapacitor electrodes thus, with good stability, exhibit very high specific capacitances and high energy densities.
 | ||
| Scheme 1 Concept and procedure for making large-area CNAGs/SMF hybrid structures. Note: a cubic architecture here is used only to demonstrate the idea and the hybrid actually shows an irregular 3D structure as illustrated by the SEM images below. | ||
| Samples | CAGs/wt (%) | CNTs/wt (%) | Ni-fiber/wt (%) | Weight ratio of CAGs to CNTs |
|---|---|---|---|---|
| CNTs/SMF-Ni | 0.0 | 50.0 | 50.0 | 0.0 |
| CNAGs/SMF-Ni-1 | 16.4 | 41.8 | 41.8 | 0.39 |
| CNAGs/SMF-Ni-2 | 21.6 | 39.2 | 39.2 | 0.55 |
| CNAGs/SMF-Ni-3 | 26.2 | 36.9 | 36.9 | 0.71 |
| CNAGs/SMF-Ni-4 | 27.6 | 36.2 | 36.2 | 0.76 |
| CNAGs/SMF-Ni-5 | 37.0 | 31.5 | 31.5 | 1.17 |
| CNAGs/SMF-Ni-6 | 42.0 | 29.0 | 29.0 | 1.44 |
| CAGs/SMF-Ni | 39.2 | 0.0 | 60.8 | ∞ |
Experimental
Materials and fabrication
A circular macroscopic CNTs/SMF-Ni composite consisting of 50 wt% CNTs was fabricated through CNT growth on a sinter-locked 3-dimensional (3D) porous SMF-Ni slab (80 mm diameter by 0.8 mm thick; consisting of 5 vol% 8 μm Ni-fibers and 95 vol% void volume) by catalytic decomposition of C2H4 in the presence of H2 at 700 °C, as previously reported elsewhere.27 The CAG was assembled on the CNTs rooted on the Ni-fiber as follows: (1) PI-sol preparation. Pyromellitic dianhydride (PMDA) of 10 mmol was dissolved in 37.7 mL N,N′-dimethylacetamide (DMAc). To the above solution was added 10 mmol of p-phenylenediamine (PDA) and then the mixture was stirred until the slurry yielded a clear yellow polyimide (PI)-sol. All chemicals (reagent grade) were purchased from Alfa Aesar and were directly used without further purification. (2) PI-sol coating of CNTs/SMF-Ni. The resulting PI-sol was used to impregnate the CNTs/SMF-Ni to allow this to coat the CNTs uniformly. The resulting sample was kept for 6 h in air at room temperature, then it was soaked overnight in acetone, followed by drying at 40 °C for 12 h. (3) Thermal treatment. The resulting PI-sol-coated CNTs/SMF-Ni composite was heated under highly purified N2 flow, by programming the temperature from 100 to 400 °C using an interval of 100 °C with a hold time of 1 h at each temperature point. This important step led to deep polymerization of PI. (4) Carbonization. Subsequently, the sample was heated from 400 to 950 °C with a ramp of 3 °C min−1 under N2 flow and then held at 950 °C for 1 h, leading to the CNAGs/SMF-Ni. By continuously repeating step 1 to step 3, a high CAG content in the whole CNAGs/SMF-Ni composite could be achieved (Table 1).Morphology, structural and textural characterization
The surface morphology and microstructure of the CNAGs/SMF-Ni samples were examined by means of scanning electron microscopy (SEM, Hitachi S-4800) operating at an accelerating voltage of 3 kV and transmission electron microscopy (TEM, JEOL-JEM-2010) operating at 200 kV. Textural and structural features were examined by N2 adsorption–desorption (Quantachrome Autosorb 3B), X-ray diffraction (XRD, Bruker Advance D8, Cu-Kα radiation (λ = 0.154056 nm)), and Raman spectroscopy (Jobin yvon T64000 equipped with a CCD multi-channel detector). The specific surface area was determined from the N2 adsorption isotherm at −196 °C using standard Brunauer–Emmett–Teller (BET) theory. Micropore surface area (Smic) and volume (Vmic), and mesopore surface area (Smes), volume (Vmes) and size distribution were analyzed by density functional theory (DFT) .Electrochemical properties studies
An electrochemical test system in 5.0 mol L−1 KOH aqueous electrolyte was built from the self-supporting CNAGs/SMF-Ni composites for both working and counter electrodes. The electrochemical characteristics of the CNAGs/SMF-Ni composites were obtained by cyclic voltammetry and galvanostatic charge–discharge techniques using a CHI-660C instrument (made in China). Impedance spectroscopy measurements were also performed using the CHI-660C.Results and discussion
Macroscopic synthesis of CNAGs/SMF-Ni composites
Fig. 1 shows SEM and TEM images for the morphology and micro-/nano-structure of the pristine CNTs/SMF-Ni and the representative CNAGs/SMF-Ni-5 composite. The resulting CNTs/SMF-Ni composite retains a macroporous 3-dimensional network structure with a uniform and dense layer of CNTs along with the Ni fiber (Fig. 1a–d). The TEM image shows that the CNTs herein showed a large hollow core, uniform diameter from 50 to 60 nm and thin wall thickness of 8–10 nm (Fig. 1d). A cross-section SEM image illustrates that the CNTs were grown along the radial direction of the 8 μm Ni-fiber core, producing a CNT layer as thick as ∼3 μm (Fig. 1c). Further closer observation by SEM shows that the CNTs, with the maintenance of their individual features, had a narrow diameter distribution and entirely opened macropores among them (Fig. 1b).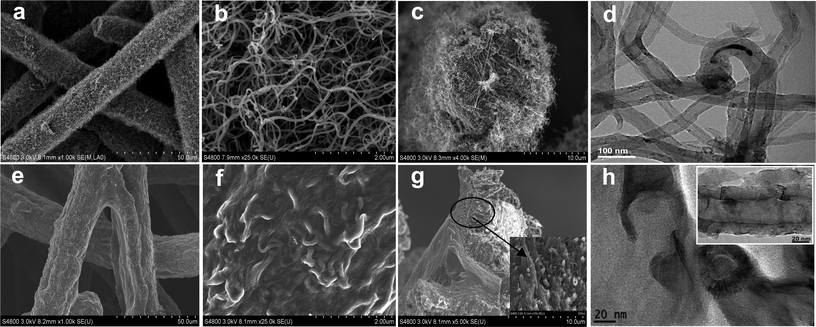 | ||
| Fig. 1 SEM and TEM images. CNTs/SMF-Ni sample: (a–c) SEM images, (d) TEM image; CNAGs/SMF-Ni-5 sample: (e–g) SEM images, (h) TEM image. | ||
After PI-sol coating and subsequent pyrolysis, the PI-derived CAGs were uniformly integrated with the CNTs rooted on the Ni fiber to form CNAGs/SMF-Ni composites while the 3-dimensional open structure was preserved very well (Fig. 1e). In brief, the void space within the CNT layer could be entirely filled with PI-sol in the impregnation step. As a result, after undergoing subsequent pyrolysis a continuous CNAG cladding along with the Ni fiber was formed and tightly fixed onto the Ni fiber through the CNTs (Fig. 1e–g). For comparison, a CAGs/SMF-Ni composite consisting of 39 wt% CAGs (equivalent to the CNAGs/SMF-Ni-5 sample in Fig. 1) was also prepared by PI-coating/pyrolysis processes. It is not surprising that carbon lumps were formed rather than continuous cladding and most of them seemed separated from the Ni-fiber surface (Fig. S1 in ESI†). The cross-section image of CNAGs/SMF-Ni shows the intrinsic inside combination structure of CAGs and CNTs (Fig. 1g). Within the CNAGs cladding, the CNTs were inserted uniformly into the CAGs agglomerate particles and connected these agglomerates firmly in their entirety (Fig. 1g). The TEM image of the CNAG composite shows the coverage of the external surface of CNTs with the PI-derived CAG fragments that consisted of a network of nanoparticles assembled in an interconnected chain-like structure (Fig. 1h and the inset). In addition, the three-dimensional network of SMF-Ni provided a basic macroporous structure, serving as the electrolyte reservoir that is helpful to reduce the ion-transfer distance from bulk electrolyte to the CNAG surface. Moreover, the self-supporting feature avoids the use of a binder for molding and permits it to be used directly for supercapacitor electrodes.
Textural and structural features of CNAGs/SMF-Ni composites
The textural and structural properties of the CNAGs/SMF-Ni composites have also been studied by N2 adsorption–desorption, XRD and Raman, with the results shown in Fig. 2, Fig. 3 and Table 2. The three representative CNAGs/SMF-Ni composites all show a type IV isotherm typical for mesoporous materials, according to the classification of the International Union of Pure and Applied Chemistry (IUPAC), as evidenced by the appearance of clear capillary condensation step in the P/P0 region of 0.45–0.50 (Fig. 2A). The composites also show a wide Barrett–Joyner–Halenda (BJH) pore size distribution while the most probable pore size is 2.1 nm (Fig. 2B). Along with the increase of CAG content in the CNAGs/SMF-Ni composites from 16.4% to 37.0%, the density of mesopores was increased clearly due to the increase of the step (P/P0 = 0.45–0.50) while the density of micropores was increased just slightly due to a small increase of the adsorbed gas amount at low relative pressures (Fig. 2A).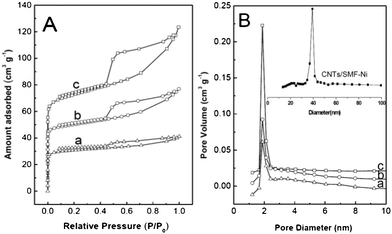 | ||
| Fig. 2 (A) N2 adsorption–desorption isotherms and (B) pore size distribution profiles of (a) CNAGs/SMF-Ni-1, (b) CNAGs/SMF-Ni-3, and (c) CNAGs/SMF-Ni-5. | ||
 | ||
| Fig. 3 XRD pattern (A) and Raman spectrum (B) of CNAGs/SMF-Ni-5 sample. | ||
| Samples | S BET/m2 g−1 | S mes /m2 g−1 | S mic/m2 g | V mic/cm3 g−1 | V mes/cm3 g | PV/cm3 g−1 | PD/nm |
|---|---|---|---|---|---|---|---|
| a BET specific surface area (SBET), micropore surface area (Smic) and volume (Vmic), mesopore surface area (Smes) and volume (Vmes), pore volume (PV) and average pore diameter (PD). | |||||||
| CNAGs/SMF-Ni-1 | 93 | 82 | 11 | 0.017 | 0.049 | 0.067 | 5.8 |
| CNAGs/SMF-Ni-3 | 130 | 95 | 35 | 0.068 | 0.073 | 0.139 | 4.3 |
| CNAGs/SMF-Ni-5 | 155 | 128 | 27 | 0.050 | 0.088 | 0.138 | 3.6 |
As noted by the inset in Fig. 2B, the CNTs/SMF-Ni composite has the characteristics of mesoporous CNTs (38 nm) with a lack of micropores, which is consistent with our previous report.27 However, pores with diameter around 38 nm are not visible in the CNAGs/SMF-Ni samples in Fig. 2B. Indeed, the SEM/TEM images above clearly indicate that the CNTs (both external wall and tube mouth) were completely covered with CAG thereby leading to the disappearance of the characteristic mesopores of CNTs. It is in turn believed that the micropores and mesopores of the CNAGs/SMF-Ni composites are all contributed by the CAGs. One can say that the CNAGs/SMF-Ni composites have a characteristic hierarchical macro-mesoporous structure with a low density of micropores (Fig. 1 and Fig. 2).
Table 2 shows some typical textural parameters of the representative composite samples. Along with the increase of the CAG amount in the CNAGs/SMF-Ni composites, their specific surface area was increased from 93 to 155 m2 g−1 whereas their average pore size was decreased from 5.8 to 3.6 nm. According to the fact that the mesopores and micropores of the CNAGs/SMF-Ni composites all come from the CAG, the specific surface area of the CAG in the composites was estimated to be 420–560 m2 g−1, to which the mesopores (pore size: > 2 nm) provided a high contribution of > 70%. In contrast, the CAGs/SMF-Ni composite delivered a specific surface area of only 90 m2 g−1 (corresponding to 230 m2 g−1 based on CAGs) and over 80% of the surface area was contributed by the micropores. For instance, the CAGs in the CNAGs/SMF-Ni-5 composite provided an estimated specific surface area of 420 m2 g−1; the CAG mesopores contributed 82.5% of the total surface area and delivered a mesopore volume of 0.24 cm3 g−1 (corresponding to 0.09 cm2 g−1 based on the overall weight of the composite). In application for carbon electrochemical capacitors, as is well known, a high mesopore surface area is an important aspect for achieving the high specific capacitance of the electrode, since the electrolyte ions nestling up to an electrode form a layer about 1 nm thick.28
For the CNAGs/SMF-Ni-5 composite, a weak and broadened XRD peak of carbon (002) was observed (Fig. 3A). The d(002) spacing was calculated to be 0.336 nm, being only slightly larger than that (0.335 nm) of the ideal graphite. The XRD results indicate that the CAG in such a composite had high graphitization but small micro-crystal size. Further insight into the structure of CNAGs/SMF-Ni-5 was obtained from Raman spectroscopy (Fig. 3B). The composite sample showed two main Raman bands centered at 1340 cm−1 (D band) and 1580 cm−1 (G band). Since Raman is a surface sensitive technique and the CNTs are covered with CAGs, it is reasonable to assign these two bands to the CAGs rather than CNTs. The G band is associated with the E2g mode (stretching vibrations) in the basal plane of graphite,29 corresponding to the graphitization degree and the graphitic plane alignment of carbon materials.29,30 The D band is sensitive to the concentration of graphite edge planes and/or crystal boundaries against standard graphite planes,18 being explained as disorder, imperfections, and finite particle size effects.29,31,32 The smaller crystalline grains and the interdefect distance produce a higher intensity ratio of the D and G bands (ID/IG) and broaden the width of the bands.29,31,32 In our case, a high ID/IG value of 1.8 was obtained for the CAGs in the CNAGs/SMF-Ni-5 composite, indicating that the CAGs are disordered. Using the empirical formula (La = 4.53IG/ID (nm)) found by Tuinstra and Koenig,29 the average basal plane diameter (La) in the graphite crystallites was estimated to be 2.5 nm. Combining this information with a CAG d(200) spacing (0.336 nm) close to the ideal graphite, it is rational to infer that the smaller crystallite grains is the main cause for the disordered feature of CAGs in the CNAGs/SMF-Ni-5 composite.
Electrochemical properties and capacitance
To demonstrate the excellent ion diffusivity and conductivity of these newly developed composites for application in supercapacitors, electrochemical impedance measurements were carried out in 5.0 mol L−1 KOH aqueous electrolyte. Fig. 4 shows the Nyquist plots, exhibiting two distinct parts including a semicircle in the high frequency region and a sloped line in the low frequency region. The charge transfer resistance (Rf) could be estimated from the semicircle diameter of the high-frequency loop, and the electrolyte ion diffusion to the mesopore was characterized by the linear part of the sloped line.33,34 The almost vertical line was obtained in the low frequency region on four CNAGs/SMF-Ni hybrid electrodes studied, clearly demonstrating a good capacitive behaviour35 without diffusion limitations that profits from their entirely accessible hierarchical macropore/mesopore structure. Ion-buffering reservoirs could be formed in the macropores so as to minimize the diffusion distances to the CNAGs, and mesopores would also permit a rapid and unimpeded diffusion of ions in the CNAG mesoporous channels. | ||
| Fig. 4 Impedance spectroscopy on the two-electrode capacitor based on CNAGs/SMF-Ni electrodes in a 5.0 mol L−1 KOH aqueous electrolyte. (a) CNAGs/SMF-Ni-2, (b) CNAGs/SMF-Ni-3, (c) CNAGs/SMF-Ni-5, (d) CNAGs/SMF-Ni-6. | ||
In the high frequency region, the semicircle became larger with the CAG content in the composites, presenting an increasing inclination indicative of the internal resistance Rf with the increase in CAGs. Nevertheless, the highest Rf was confirmed to be only ∼0.8 Ω for CNAGs/SMF-Ni-6 with the highest CAG content of 42 wt%, whereas the CAGs/SMF-Ni hybrid without CNTs delivered a large internal resistance of ∼3.0 Ω. This clearly indicates the excellent conductivity of our CNAGs/SMF-Ni hybrid electrodes, mostly due to their unique integrated structure in which the Ni-fiber network serves as current collector and the CNTs grown on the Ni-fiber act as nano conducting wires to link the charge-storage CAGs.
The electrochemical energy storage performance of the CNAGs/SMF-Ni composite electrodes was evaluated by cyclic voltammogram (CV) and galvanostatic charge–discharge measurements in a 5.0 mol L−1 KOH electrolyte, using a three-electrode system where the composite served as both the working and the auxiliary electrodes. Fig. 5 shows the CV curves of the supercapacitor with various working electrodes versus Hg/Hg2SO4 (SCE) at a high potential scan rate of 100 mV s−1. By taking advantage of the accessible macro-/meso-porous structure and good conductivity, it is not surprising that the composite electrodes, including CNAGs/SMF-Ni-6 with a very high CAG content of 42 wt%, all presented typical “box-like” shapes even at a high scan rate of 100 mV s−1. This indicates an ideal double-layer capacitor with quick dynamics of charge propagation,17 being consistent with the observation of the vertical line in the low frequency region of the Nyquist plots (Fig. 4). Fig. 6 shows the charge–discharge profiles for the CNAGs/SMF-Ni-5 composite electrode measured using different current densities. Typical triangular curves were retained almost with no rapid voltage change at the initial stage of the charge and the discharge, indicating again the characteristic of double layer charge storage mechanism. Another noticeable feature of the charge–discharge curves was the very small ohmic drop when the current sign was reversed even at a current density of 500 mA g−1, indicating a low value of resistance of the electrodes. Moreover, a specific capacitance of 348 F g−1 was obtained from galvanostatic discharge at a current density of 200 mA g−1, being compatible with that from the CV data.
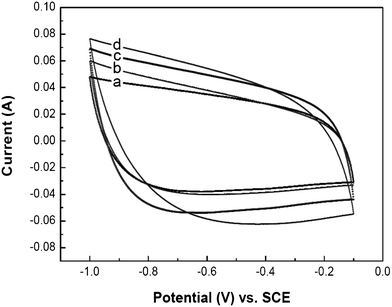 | ||
| Fig. 5 Cyclic voltammograms of as-made CNAGs/SMF-Ni composites for both working and counter electrodes at a scan rate of 100 mV s−1 in a 5.0 mol L−1 KOH aqueous electrolyte. (a) CNAGs/SMF-Ni-2, (b) CNAGs/SMF-Ni-3, (c) CNAGs/SMF-Ni-5, (d) CNAGs/SMF-Ni-6. | ||
 | ||
| Fig. 6 Charge–discharge curves of CNAGs/SMF-Ni-5 electrode vs. current densities in a 5.0 mol L−1 KOH aqueous electrolyte. | ||
Table 3 summarizes the features of the CNAGs/SMF-Ni electrode based supercapacitors. The specific capacitances were calculated by the CV method using the following equation:

| Samples | Capacitance (F g−1) at various scan rates (mV s−1) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 5 | 10 | 100 | |
| a The specific capacitance data are all on the basis of CAGs except for CNTs/SMF-Ni (based on CNTs). | |||||
| CNTs/SMF-Ni | 79 | 65 | 60 | 56 | 47 |
| CNAGs/SMF-Ni-1 | 232 | 226 | 217 | 201 | 165 |
| CNAGs/SMF-Ni-2 | 249 | 232 | 226 | 211 | 176 |
| CNAGs/SMF-Ni-3 | 278 | 264 | 248 | 234 | 195 |
| CNAGs/SMF-Ni-4 | 319 | 307 | 289 | 277 | 225 |
| CNAGs/SMF-Ni-5 | 359 | 334 | 321 | 301 | 260 |
| CNAGs/SMF-Ni-6 | 340 | 329 | 307 | 298 | 240 |
| CAGs/SMF-Ni | 53 | 50 | 48 | 45 | 36 |
The CNTs-free CAGs/SMF-Ni hybrid electrode was also tested and delivered a very small specific capacitance of only 45 F g−1 at a scan rate of 10 mV s−1. In contrast, the CNAGs/SMF-Ni-5 electrodes provided a capacity (301 F g−1 at 10 mV s−1 scan rate) >5-fold higher than that for the CAGs/SMF-Ni electrodes. This essential difference is clearly assignable to the discrepancy in domain microstructure between these two hybrid electrodes. Actually, unlike the CNAGs/SMF-Ni-5 hybrid which has CNTs to reinforce the CAGs to form uniform CNAGs and meanwhile to fix them firmly onto the current collector Ni-fibers (Fig. 1e–g), the CAGs in the CAGs/SMF-Ni hybrid were full of cracks and were split from the Ni-fibers (Fig. S1 in ESI†). Regarding the CNTs/SMF-Ni hybrid, the specific capacitance on the basis of the mass of CNTs was about 56 F g−1 at a scan rate of 10 mV s−1, being consistent with our previous report.27 Nevertheless, it is believed that CNTs in the CNAGs/SMF-Ni hybrid electrodes might provide very little or no contribution to their capacitance, due to the fact that the CNTs were entirely covered with the CAGs (Fig. 1). In addition, the hybrid electrodes all showed a relatively small decrease indicative of the specific capacitances by ∼30%, along with the increase of the potential scan rates from 1 to 100 mV s−1. However, activated carbon-based electrodes provide significant degradation as the potential scan rate is increased.36 These results suggest that our hybrid electrodes have good rate capability, which is very important for the electrode materials of a supercapacitor to provide high power density. Undoubtedly, our novel strategy is working successfully to result in high-performance supercapacitor electrodes through nano current wire CNTs to combine the charge storage CAGs and current collector Ni-fibers.
The cyclability testing was performed on the CNAGs/SMF-Ni-5 electrode by continuous CV measurement in a 5.0 mol L−1 KOH aqueous electrolyte at a scan rate of 100 mV s−1, with the results as shown in Fig. 7. Through entire the 1000 CV cycles, the CV curves were always characteristic of the typical “box-like” shape (inset in Fig. 7), indicating the ideal double layer charge storage feature. The specific capacitance presented a continuous slow decrease in the initial 300 cycles and then remained stable throughout the test. About 95% specific capacitance was retained after 1000 CV cycles, predicting a good durability of our novel hybrid electrodes.
 | ||
| Fig. 7 Cyclability of CNAGs/SMF-Ni-5 electrode in a 5.0 mol L−1 KOH aqueous electrolyte. Data were obtained from cyclic voltammogram testing results using a scan rate of 100 mV s−1. | ||
Conclusions
On the basis of a novel sinter-locked 3-dimensional network consisting of 5 vol% 8 μm Ni fibers and 95 vol% void volume, thin-sheet Ni-fiber-supporting carbon nanotubes (CNTs) and carbon aerogel (CAG) hybrid electrodes, namely CNAGs/SMF-Ni, have been successfully fabricated for supercapacitors on a macroscopic scale by the following procedures. Firstly, a thin-sheet CNTs/SMF-Ni hybrid with 50 wt% CNTs was prepared by the catalytic chemical vapor deposition of ethylene on the microfibrous structure using Ni fibers. Secondly, CAGs up to 42 wt% were assembled onto the CNTs rooted into the microfibrous network by polyimide-sol coating/pyrolysis technology to create new structural self-supporting hybrid electrodes in which a Ni microfiber network serves as current collector, CNTs as nano conducting wire and CAGs as ion storage reservoir. This approach permits the desired macroscopic fabrication of CNAGs-based self-supporting electrodes in a simple and cost-effective way and provides a unique combination of binderlessness, excellent electric conductivity, hierarchically porous structure and large mesopore surface area. Owing to the excellent ion diffusivity, high electric conductivity and large mesopore surface area, high specific capacitances (e.g., 359 F g−1 at a scan rate of 1 mV s−1) could be obtained in a 5.0 mol L−1 KOH aqueous electrolyte solution. Moreover, in 1000 CV testing cycles, the CNAGs/SMF-Ni-5 (CAGs content: 37 wt%) hybrid electrode, with a good stability, showed a very small decrease of the specific capacitance of only 5% after the initial 300 cycles and then remained constant until the end of the test.Acknowledgements
This work was funded by the NSF of China (21076083, 20973063), the “973 program” (2011CB201403) from the MOST of China, the Shanghai Rising-Star Program (10QH1400800), the Specialized Research Fund for the Doctoral Program of Higher Education (20090076110006), the Shanghai Leading Academic Discipline Project (B409), and the Guangdong Industry-University Joint Research Program (2011B090400389).References
- (a) D. S. Su and R. Schlögl, ChemSusChem, 2010, 3, 136 CrossRef CAS; (b) S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972 RSC; (c) S.-K. Park, S.-H. Yu, N. Pinna, S. Woo, B. Jang, Y.-H. Chung, Y.-H. Cho, Y.-E. Sung and Y. Piao, J. Mater. Chem., 2012, 22, 2520 RSC; (d) P. Johns, M. Roberts and J. Owen, J. Mater. Chem., 2011, 21, 10153 RSC.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS.
- C. G. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863 CrossRef CAS.
- G. Lota, K. Fic and E. Frackowiak, Energy Environ. Sci., 2011, 4, 1592 CAS.
- S. Roldán, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Angew. Chem., Int. Ed., 2011, 50, 1699 CrossRef.
- S.-L. Chen, H.-Q. Hou, F. Harnisch, S. A. Patil, A. A. Carmona-Martinez, S. Agarwal, Y.-Y. Zhang, S. Sinha-Ray, A. L. Yarin, A. Greiner and U. Schröder, Energy Environ. Sci., 2011, 4, 1417 CAS.
- S. H. Aboutalebi, A. T. Chidembo, M. Salari, K. Konstantinov, D. Wexler, H. K. Liu and S. X. Dou, Energy Environ. Sci., 2011, 4, 1855 CAS.
- J. C. Lytle, J. M. Wallace, M. B. Sassin, A. J. Barrow, J. W. Long, J. L. Dysart, C. H. Renninger, M. P. Saunders, N. L. Brandell and D. R. Rolison, Energy Environ. Sci., 2011, 4, 1913 CAS.
- X. Liu, Y. S. Hu, J. O. Müller, R. Schlögl, J. Maier and D. S. Su, ChemSusChem, 2010, 3, 261 CrossRef CAS.
- Z.-B. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866 CAS.
- H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277 CAS.
- Z.-Q. Niu, W.-Y. Zhou, J. Chen, G.-X. Feng, H. Li, W.-J. Ma, J.-Z. Li, H.-B. Dong, Y. Ren, D. Zhao and S.-S. Xie, Energy Environ. Sci., 2011, 4, 1440 CAS.
- Z.-P. Chen, W.-C. Ren, L.-B. Gao, B.-L. Liu, S.-F. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS.
- X. C. Zhao, A. Q. Wang, J. W. Yan, G. Q. Sun, L. X. Sun and T. Zhang, Chem. Mater., 2010, 22, 5463 CrossRef CAS.
- H. Q. Li, R. L. Liu, D. Y. Zhao and Y. Y. Xia, Carbon, 2007, 45, 2628 CrossRef CAS.
- F. Béguin, K. Szostak, G. Lota and E. Frackowiak, Adv. Mater., 2005, 17, 2380 CrossRef.
- T. Bordjiba, M. Mohamedi and L. H. Dao, Adv. Mater., 2008, 20, 815 CrossRef CAS.
- C. Peng, S.-W. Zhang, D. Jewell and G. Z. Chen, Prog. Nat. Sci., 2008, 18, 777 CrossRef CAS.
- R. W. Pekala, J. C. Farmer, C. T. Alviso, T. D. Tran, S. T. Mayer, J. M. Miller and B. Dumn, J. Non-Cryst. Solids, 1998, 225, 74 CrossRef CAS.
- T. Horikawa, J. Hayashi and K. Muroyama, Carbon, 2004, 42, 169 CrossRef CAS.
- V. E. Yudin, M. Y. Goykhman, K. Balik, P. Glogar, P. Polivka, G. N. Gubanova and V. V. Kudryavtsev, Carbon, 2002, 40, 1427 CrossRef CAS.
- T. Takeichi, Y. Yamazaki, M. Zuo and A. Ito, Carbon, 2001, 39, 257 CrossRef CAS.
- T. Takeichi, Y. Eguchi, Y. Kaburagi, Y. Hishiyama and M. Inagaki, Carbon, 1999, 37, 569 CrossRef CAS.
- T. Bordjiba, M. Mohamedi and L. H. Dao, J. Power Sources, 2007, 172, 991 CrossRef CAS.
- T. Bordjiba, M. Mohamedi and L. H. Dao, Nanotechnology, 2007, 18, 035202 CrossRef.
- F. T. Jiang‚, Y. Z. Fang‚, Y. Liu‚, L. Chen‚, Q. S. Xue‚, Y. Lu‚, J. X. Lu and M. Y. He, J. Mater. Chem., 2009, 19, 3632 RSC.
- R. F. Service, Science, 2006, 313, 902 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus and A. Jorio, J. Phys. Chem. C, 2007, 111, 17887 CAS.
- N. Soin, S. S. Roy, C. O'Kane, J. A. D. McLaughlin, T. H. Lim and C. J. D. Hetherington, CrystEngComm, 2011, 13, 312 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2001, 64, 075414 CrossRef.
- D. W. Wang, F. Li, H. T. Fang, M. Liu, G. Q. Lu and H. M. Cheng, J. Phys. Chem. B, 2006, 110, 8570 CrossRef CAS.
- W. Sugimoto, H. Iwata, K. Yokoshima, Y. Murakami and Y. Takasu, J. Phys. Chem. B, 2005, 109, 7330 CrossRef CAS.
- Y. Wang, Z. Q. Shi, Y. Huang, Y. F. Ma, C. Y. Wang, M. M. Chen and Y. Y. Chen, J. Phys. Chem. C, 2009, 113, 13103 CAS.
- G. Lota, T. A. Centeno, E. Frackowiak and F. Stoeckli, Electrochim. Acta, 2008, 53, 2210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. See DOI: 10.1039/c2ra20271a |
| This journal is © The Royal Society of Chemistry 2012 |