Selective reduction of visible upconversion emissions induced by Bi3+ in Tm3+/Yb3+-doped Y0.89−xBixVO4 microcrystals†
Chanchal
Hazra
,
Shyam
Sarkar
and
Venkataramanan
Mahalingam
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER), Kolkata, Mohanpur, West Bengal, India. E-mail: mvenkataramanan@yahoo.com; Fax: +91-33-25873020; Tel: 91-0-9007693474
First published on 24th May 2012
Abstract
In this article we have shown that the intensity ratio of near infrared (NIR) to blue upconversion emission from Tm3+ ions can be enhanced up to 800 times by simple control of the concentrations of Bi3+/Y3+ in Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 (x = 0 to 0.89) microcrystals. The intensity of the strong blue emission occurring near 475 nm due to the 1G4→3H6 transition is selectively reduced upon bismuth doping, while the intensity of the NIR emission at 800 nm (3H4→3H6) is hardly affected. The enhanced NIR/blue emission intensity achieved via the upconversion process is advantageous in reducing the background scattering, and consequently increases the contrast in the bio-imaging applications. Several control experiments were performed to unravel the role of Bi3+ ions in the reduction of the blue emission intensity. An energy transfer mechanism involving Tm3+, Bi3+ and Yb3+ ions is proposed for the preferential reduction in intensity of the blue emission. All microcrystal samples were completely characterized using XRD, Raman and photoluminescence methods.
Introduction
Lanthanide (Ln3+)-doped materials have several applications, such as in the development of new phosphors for display and lighting, laser crystals and bio-markers, to name just a few.1 One of the interesting characteristics of Ln3+ ions is their sharp transitions arising due to intra 4f transitions, which are shielded by the outer 5s and 5p orbitals.2 Despite the fact that these transitions have low absorption coefficients due to forbidden parity, the long lifetimes possessed by the Ln3+ ions are exploited for several optoelectronic applications.3 In addition to the Stokes shifted luminescence, they possess the ability to exhibit anti-Stokes emissions, which is generally known as the upconversion process.4The upconverting properties of the lanthanide ions have been widely studied in various matrices, but oxide-based materials dominate the literature.5 This is because they are very robust, possessing a high melting temperature, and can easily be prepared by several simple methods like sol–gel, combustion and precipitation followed by solid state synthesis.6 Moreover, most of the lanthanide ions exhibit strong optical signals in oxide-based matrices such as Y2O3, Gd2O3, ZrO2, BaTiO3, etc.7
Further enhancement of the Ln3+ emissions has also been observed in oxide-based materials by doping with Li+ or Bi3+ ions. For example, the Eu3+ emissions were enhanced in Gd2O3:Eu3+ thin films by additional doping of Li+ into the matrix.8 Though the exact role of Li+ in enhancing the emission intensities is not well resolved, it is broadly believed to happen via a reduction in symmetry around the Ln3+ ions, thereby relaxing the selection rules.9 Similarly, quite a few reports are available on the enhancement of Ln3+ emissions, particularly of Eu3+, by additional doping of Bi3+ ions into oxide materials.10 A small amount of Bi3+ doping (0.2 mol %) in GdVO4:Eu3+ led to improved Eu3+ emissions.11 Enhanced red emission was also reported in Bi3+-doped CaMoO4:Eu3+.12 Unlike Li+, the increase in emission intensity by Bi3+ doping is ascribed to sensitization via energy transfer (ET), where the energy is transferred from the 1P1 excited state of Bi3+ to Eu3+ excited energy levels.10 However, incorporation of either Li+ or Bi3+ generally enhances all the emission signals, but no selectivity is observed.
Selective increase or decrease in the intensity of one or a few peaks is interesting, as it would lead to a very characteristic emission pattern with specific signals, which can be used as a bio-marker or in authentication applications. For example, the Tm3+/Yb3+-doped materials typically exhibit two strong emissions, one near 475 nm, due to the 1G4→3H6 transition, while the other one is in the NIR region close to 800 nm (3H4→3H6).13 Reducing the blue emission in the visible region would increase the NIR to blue emission ratio and result in a unique emission pattern. Moreover, an increase in the NIR to blue emission ratio is important for enhancing the contrast of the imaging process, as NIR is silent to tissues.14,15 In fact, we have recently shown preferential suppression of the high energy emissions of Tm3+ emission by Dy3+ ions in Tm/Yb/Dy-doped LiYF4 microcrystals, whereas the NIR emission at 800 nm was less affected up to a certain Dy3+ concentration.16
In this paper we have shown that by simple control of the Bi3+/Y3+ concentration in Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 (x = 0 to 0.89) solid solutions, the upconverted emission intensity ratio of NIR to blue can be well tuned. A reduction in the NIR to blue upconversion emission intensity of about 800 times has been noted by completely replacing Y3+ ions by Bi3+ ions. This is achieved by reducing the intensity of the blue emission without affecting the NIR emission. Several control experiments were performed to unravel the mechanism and it was found that Bi3+ ions play a major role in the reduction of blue emission from the Tm3+ ions in Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 (x = 0 to 0.89) microcrystals. The selective reduction in the intensities of the visible emissions (blue and red) without affecting the NIR peak intensity is interesting for several applications such as in biology, authentication, etc.
Experimental procedure
The Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals (x = 0 to 0.89) were prepared via the Pechini-type sol–gel method. Briefly, the stoichiometric amounts of Y2O3, Yb2O3 and Tm2O3 were converted into their corresponding nitrates by dissolving in 1 M HNO3, whereas for the Bi source Bi(NO3)3·5H2O was used as received. After completely dissolving, 1 mmol NH4VO3 was added to the above mixture under stirring to obtain a yellow coloured solution followed by the addition of 2 mol citric acid. Subsequently, 4 g polyethylene glycol (PEG-8000) was added, and the mixture continued to be stirred for another 30 min before being transferred to the crucible and left in the oven at 90 °C for 24 h. The resulting gel was placed inside a high temperature furnace (Lindberg) and heated to 800 °C for 16 h at a rate of 3 °C per minute. The sample was maintained at that temperature for 16 h before cooling down to room temperature at a rate of 3 °C per minute. All the samples discussed in the text were prepared under identical conditions following the same protocol.The XRD patterns were collected using a Rigaku-SmartLab diffractometer attached with a D/tex ultra detector and a Cu-Kα source operating at 50 mA and 40 kV. The scan range was set from 10–90° 2θ with a step size of 0.02° and a count time of 2 s. The samples were well powdered and spread evenly on a quartz slide. The Raman measurements were performed using a Horiba Jobin Yvon LabRAM HR 800 micro-Raman spectrometer, using a 1800 g mm−1 grating. The samples were excited with a 633 nm He–Ne laser line. Field emission-SEM images were collected on a JEOL, JSM-6700F, Japan. Prior to loading the samples into the chamber, they were coated with a thin film of gold in order to avoid charging effects. The photoluminescence spectra were measured on a Horiba Jobin Yvon spectrometer equipped with a 150 W Xe lamp. The nanocrystal powders were well ground before being evenly spread on a solid sample holder. The excitation and emission light were dispersed using a Czerny-Turner monochromator with an optical resolution of 1 nm. The emitted photons were detected using a Hamamatsu R928 detector. The output signal was recorded using a computer. For the upconversion measurements, a 980 nm diode laser from RgBLase LLC, which was coupled with a fibre, with a core diameter of 100 microns, was used as the excitation source. The output signal was measured with the Jobin Yvon spectrometer as detailed above. The lifetime measurements were done by exciting the sample at 270 nm using a pulsed Xe lamp, and measuring the blue emission at 475 nm.
Results and discussion
The sol–gel method used for the synthesis of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals led to the formation of microcrystals, as evidenced from the SEM images of samples with x = 0 and 0.89 (Fig S1 in the ESI†). The concentration of the Tm3+ and Yb3+ ions (1 and 10%) is fixed for all the samples. The X-ray diffraction (XRD) patterns of a series of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals (x = 0 to 0.89), prepared via a Pechini type sol–gel method, are displayed in Fig. 1. First, the pattern obtained for the Tm3+/Yb3+-doped YVO4 sample (i.e. without bismuth) matches well with the tetragonal phase YVO4 structure, except for a weak peak at the 2θ value 29°, which could be assigned to the (222) plane of Y2O3. Upon Bi3+ doping, the formation of the tetragonal phase is preserved, as is apparent from the XRD patterns up to 25% Bi3+ doping (x = 0.25). This suggests the formation of solid solutions of the structure Y0.89−x(BixTm 0.01Yb0.1)VO4, until x reaches 0.25. For microcrystal samples with higher Bi3+ concentration (x = 0.50 and 0.75), the peak at 25° is shifted towards lower 2θ. This shift is likely due to the difference in the ionic radii between Bi3+ and Y3+, the latter being slightly smaller than the former. In addition, a sharp peak that started to appear near 28.5° is characteristic of the monoclinic BiVO4 phase. However, the tetragonal phase is still dominant, as is clear from the appearance of a strong peak at 24.5°. The formation of a predominant monoclinic phase is noticed for the Tm3+/Yb3+-doped BiVO4 microcrystals. A similar result has been observed for BixLn1-xVO4 (Ln![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Eu to Gd) bulk phosphors, where formation of the tetragonal phase is observed for x < 0.65, while for higher Bi3+ doping, the monoclinic phase formation was noticed.17
Eu to Gd) bulk phosphors, where formation of the tetragonal phase is observed for x < 0.65, while for higher Bi3+ doping, the monoclinic phase formation was noticed.17
 | ||
| Fig. 1 XRD patterns of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals, along with the standard patterns of both tetragonal and monoclinic bismuth vanadate. | ||
The XRD results are further substantiated by Raman analysis. Moreover, the latter being more sensitive than XRD, more information about the phase can be obtained. The Raman spectra (100 to 1000 cm−1) of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals (x = 0 to 0.89) are shown in Fig. 2. It is quite clear that the YVO4 sample without Bi3+ doping is tetragonal. The peaks appearing at 892, 839, 815.7 and 380 cm−1 are ascribed to the internal vibrational modes of the [VO4]3− anion.18 The first three peaks are assigned to the V–O symmetric stretching vibrations (υs) whereas the peak at 380 cm−1 is attributed to the bending vibrations (δs) of the VO43− group. The other bands appearing near 260 and 160 cm−1 are associated with external modes, which are generally symmetry related.19 There is hardly any change in the Raman spectra of Y0.89−xBixVO4 microcrystal samples up to x = 0.25 Bi3+ doping, except for a decrease in the intensity of the peaks at 892 and 260 cm−1 with increasing Bi3+ concentration. This clearly suggests that there is no major structural change as the tetragonal phase is retained. Further increase in the Bi3+ doping level leads to broadening of the Raman bands. Furthermore, a broad peak at 812 cm−1 and some new peaks near 123 cm−1 and 204 cm−1 appear. These peaks are characteristic of the formation of the monoclinic phase of BiVO4.18,20 Moreover, a broad shoulder appears on the high frequency side of the 812 cm−1 peak, indicating that a small amount of the tetragonal phase might still be present. Although the broadening of the Raman peak can be ascribed to the formation of the amorphous phase, we believe that this is less likely, as the synthesis was performed at a high temperature (800 °C), which generally enhances the crystallinity of the materials. It is quite clear from both XRD and Raman analyses that a tetragonal to monoclinic phase started to appear upon increasing the concentration of Bi3+ in Tm3+/Yb3+-doped Y0.89−xBixVO4 microcrystals.
 | ||
| Fig. 2 Raman spectra of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 (x = 0 to 0.89) microcrystals. | ||
The optical properties of the Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals (x = 0 to 0.75) display interesting phenomena. The upconversion emission spectrum of Tm/Yb-doped YVO4 microcrystals (without Bi3+), obtained by excitation with a 980 nm diode laser, is shown in Fig. 3. The spectrum displays an intense blue emission near 475 nm, weak red emissions centered at 650 and 700 nm and a strong NIR emission close to 800 nm. These peaks are quite characteristic of Tm3+ ions and were assigned to the 1G4→3H6, 2F2,3→3H6 and 3H4→3H6 transitions, respectively. These transitions occurred via energy transfer from Yb3+ ions, which are used as sensitizers for Tm3+ ions, as the Yb3+ ions possess a higher absorption coefficient at 980 nm.21 To understand the mechanism of the upconversion process, we have performed power dependent studies. The laser power (P) is related to the number of photons involved in producing the upconversion emission via the equation P α In, where I is the intensity of the upconversion emission and n is the number of photons. A logarithmic plot of P versus I will lead to the slope, which is nothing but n. The calculated values are 2.3, 1.77 and 1.7 for the 475, 650 and 802 nm emissions, respectively (see Fig. S2† for the graph). This suggests the involvement of 3, 2 and 2, 980 nm photons in producing blue, red and NIR emissions. The observed lower value is due to the saturation of the upconversion process, which has been reported earlier.22 A schematic of the energy transfer mechanism is illustrated in the inset of Fig. 3
 | ||
| Fig. 3 Upconversion emission spectrum of Tm3+(0.01)/Yb3+(0.10)-doped YVO4 microcrystals. The inset shows the schematic of the energy transfer mechanism involving Yb3+ and Tm3+ ions. | ||
The upconversion emission spectra for the Bi3+-doped samples are displayed in Fig. 4. Upon doping Bi3+ into the Tm3+/Yb3+-doped YVO4 microcrystals, the intensity of the blue emission at 475 nm is gradually decreased until x = 0.25, but for higher Bi3+ concentration (x > 0.50) a larger reduction in the emission intensity is observed. Furthermore, the intensity of the red emission is also reduced but to a lesser extent. Interestingly, the NIR emission at 800 nm due to the 3H4→3H6 transition is hardly affected. To eliminate any effect of scattering on the emission intensity between different samples, we have considered only the intensity ratio between the NIR and blue emission peaks. Thus, the calculated NIR/blue emission intensity is plotted against the Bi3+ concentration and shown in the inset of Fig. 4. The graph indicates a significant reduction (∼800 times) in the blue emission intensity with respect to the NIR emission at 802 nm by completely replacing Y3+ by Bi3+ in the Y0.89−xBixVO4 microcrystals. It is clear from the graph that there is a steady decrease in the intensity of the blue emission up to x = 0.75 Bi3+ doping, and from then on it is quite steep. At first sight, it appears as though a tetragonal to monoclinic phase transition might be the reason for the changes in the optical properties, due to relaxation of the symmetry rules. However, we strongly presume that this may not be the reason, as such a process would enhance the emission intensity rather than decrease it, as the monoclinic phase has less symmetry than the tetragonal phase. To elucidate the reason for the observed change in the optical characteristics induced upon bismuth doping, a few control experiments were performed.
 | ||
| Fig. 4 Upconversion emission spectra of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals. Inset illustrates the increase in NIR/blue emission intensity with the increase in Bi3+ concentration with the error bars. | ||
First, to ascertain that the observed changes in the emission pattern are not due to the sample color (yellowish-brown), we measured the absorption spectrum. The absorbance spectra of both the Tm/Yb-doped BiVO4 as well as the YVO4 microcrystal samples are displayed in Fig. S3 (supplementary information†). A strong absorption was observed close to 280 nm for both the samples, which is assigned to the VO43− absorption. The absorption spectra are quite similar in the visible region, except for a weak hump around 330 nm for the YVO4 sample. To verify further, we have prepared Er/Yb-doped BiVO4 microcrystals under similar experimental conditions. The reason for doing this experiment is that Er and Yb show green emission upon upconversion with a 980 nm laser. Indeed, we observed a strong green emission typical of Er3+ ions. This clearly ruled out the possibility of an internal filter effect by the BiVO4 microcrystals. The upconversion emission spectrum along with the digital image of the green emission is shown in Fig. S4 of the ESI.†
Raman and XRD analysis of Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals (x = 0 to 0.89) indicate a possible phase separation at a higher Bi3+ concentration. To verify whether the observed decrease in the blue emission intensity is due to phase separation, we prepared Tm3+-doped YbVO4 (no Bi ions) under identical experimental conditions. The upconversion emission spectrum measured from this sample is shown in Fig. 5. It is very clear that the Tm3+(0.01)-doped YbVO4 microcrystal sample exhibits a strong blue emission. To further understand the role of the Bi ions, we have prepared Tm3+-doped Yb0.89−xBixVO4 microcrystals with x = 0.05, 0.1, and 0.25. The reason for limiting the Bi doping to 25% is because a higher Bi3+ concentration might lead to phase separation, as indicated by Raman as well as XRD (vide supra). The upconversion emission spectra measured from these samples clearly indicate a significant decrease in blue emission intensity (Fig. 5). The absence of any phase separation upon additional Bi doping in Tm3+-doped YbVO4 microcrystals is confirmed by XRD and Raman analyses, which are shown in Fig. S5 and S6 of the ESI.† The above results clearly point out that Bi3+ doping alters the upconversion emissions of the Tm3+ ions, particularly the blue emission occurring near 475 nm originating from the 1G4 level.
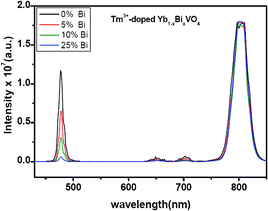 | ||
| Fig. 5 Upconversion emission spectra of Tm3+(0.01)-doped Yb0.89−xBixVO4 microcrystals. | ||
To get further insight, the emission analysis was performed by exciting the Bi3+-doped YbVO4 samples in the UV region. Moreover, the direct excitation of Tm3+ avoids any sensitization effect by Yb3+ ions. The samples were excited at 270 nm, which matches in energy with the 3PJ level of the Tm3+ ions. It is clear from the emission spectra (Fig. 6) that the blue emission is again decreased in energy with the increase in the concentration of Bi3+ ions. This clearly suggests an energy transfer taking place from the Tm3+ to the Bi3+ ions.
 | ||
| Fig. 6 Emission spectra of Tm3+(0.01)-doped Yb0.89−xBixVO4 microcrystals. | ||
There are many reports available on the use of Bi3+ as a sensitizer for Ln3+ ions, mostly for the Eu3+ ions.10 The ground state of Bi3+ is 1S0, while the excited states are 1P1, 3P2, 3P1and 3P0 (decreasing energy). The 3P0 level is placed below the 3P1 level and is very close to it in energy. The allowed transitions are 1S0→1P1 and 3P1, with broad absorption and emission signals due to its band structure.23 The transition from 1P1 to the ground state generally occurs in the UV region, whereas the location of the emission band originating from 3P1,0 can be anywhere from 380 nm to 650 nm.24 A large Stokes shift has been observed for these bands, depending on the matrix type. For example, Steler and Srivastava have reported the positions of the 3P1,0 band in pyrochlore materials.23 They found that in Sr2GdGaO5 the band was observed near 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (476 nm). In Sm/Bi-codoped Gd2O3, the 3P1,0→1S0 emission is observed near 500 nm. It is reasonable to presume from the above reports that the energy level of the 3P1,0 band is closer or lower in energy compared to the 1G4 level of the Tm3+ ions, allowing an overlap between the orbitals for energy transfer from the Tm3+ to the Bi3+ level. However, we believe this overlap may not be very efficient, and necessitates a higher Bi3+ concentration. The proposed energy transfer from the Tm3+ to the Bi3+ level should result in emission from the Bi3+ excited states. However, we do not observe any emission related to Bi3+ ions. The absence of any Bi3+ related emission is attributed to the following reasons. Firstly, the excited energy is decayed non-radiatively to the 3P0 level, as the 3P0 level is placed below the 3P1 energy level and the difference in energy may require only a few VO43− vibrations to match. The emission from 3P0 to the ground state is generally forbidden. In addition, the absence of Bi emission may be due to concentration quenching as well. This is a reasonable prediction, as the Bi3+ concentration is quite high in the matrix and the onset of concentration quenching is even lower than 5%.22 The absence of any Bi3+ emission even under UV excitation (270 nm) supports the fact that the Bi3+ emission is quenched at higher Bi3+ concentrations (see Fig. 6). Additionally, it is clear that the observed decrease in the intensity of the blue emission is similar to that noticed for Tm/Yb-doped Y0.89−xBixVO4 microcrystals via the upconversion process. This suggests that Bi3+ doping plays a role in decreasing the intensity of the blue emission from the 1G4 level of the Tm3+ ions. This fact is supported by the decrease in the lifetime of the 1G4 level of Tm3+ upon increasing the concentration of the bismuth ions in the matrix, suggesting additional relaxation channels, presumably through 3p levels of Bi3+ ions, which are closer in energy. For an undoped sample, a value of 107 μs is observed, which is reduced to 10 μs for complete replacement of Y3+ by Bi3+ ions in Tm/Yb-doped Y0.89−xBixVO4 microcrystals. The decay curves, along with the instrument response function, are shown in Fig. S7.† The other reason involves a transfer of excited Bi3+ energy into the 2F7/2 level of Yb3+ ions. Huang and Zhang observed a similar energy transfer in Bi/Yb-doped Gd2O3 phosphors, where they observed an increase in the NIR emission (980 nm) of Yb3+ ions when the sample was excited in the UV region.22a They have attributed the result to the transfer of Bi3+ 3P1 energy to two Yb3+ ions via the downconversion process. We emphasize that it is likely that the change in optical properties can be attributed to the change in the crystal phase, as has been observed in the TiO2 and NaYF4 matrices.25 However, we believe that with only a slight difference in energy levels between tetragonal and monoclinic BiVO4, it is hard to attribute the observed specific reduction in the blue emission intensity.
000 cm−1 (476 nm). In Sm/Bi-codoped Gd2O3, the 3P1,0→1S0 emission is observed near 500 nm. It is reasonable to presume from the above reports that the energy level of the 3P1,0 band is closer or lower in energy compared to the 1G4 level of the Tm3+ ions, allowing an overlap between the orbitals for energy transfer from the Tm3+ to the Bi3+ level. However, we believe this overlap may not be very efficient, and necessitates a higher Bi3+ concentration. The proposed energy transfer from the Tm3+ to the Bi3+ level should result in emission from the Bi3+ excited states. However, we do not observe any emission related to Bi3+ ions. The absence of any Bi3+ related emission is attributed to the following reasons. Firstly, the excited energy is decayed non-radiatively to the 3P0 level, as the 3P0 level is placed below the 3P1 energy level and the difference in energy may require only a few VO43− vibrations to match. The emission from 3P0 to the ground state is generally forbidden. In addition, the absence of Bi emission may be due to concentration quenching as well. This is a reasonable prediction, as the Bi3+ concentration is quite high in the matrix and the onset of concentration quenching is even lower than 5%.22 The absence of any Bi3+ emission even under UV excitation (270 nm) supports the fact that the Bi3+ emission is quenched at higher Bi3+ concentrations (see Fig. 6). Additionally, it is clear that the observed decrease in the intensity of the blue emission is similar to that noticed for Tm/Yb-doped Y0.89−xBixVO4 microcrystals via the upconversion process. This suggests that Bi3+ doping plays a role in decreasing the intensity of the blue emission from the 1G4 level of the Tm3+ ions. This fact is supported by the decrease in the lifetime of the 1G4 level of Tm3+ upon increasing the concentration of the bismuth ions in the matrix, suggesting additional relaxation channels, presumably through 3p levels of Bi3+ ions, which are closer in energy. For an undoped sample, a value of 107 μs is observed, which is reduced to 10 μs for complete replacement of Y3+ by Bi3+ ions in Tm/Yb-doped Y0.89−xBixVO4 microcrystals. The decay curves, along with the instrument response function, are shown in Fig. S7.† The other reason involves a transfer of excited Bi3+ energy into the 2F7/2 level of Yb3+ ions. Huang and Zhang observed a similar energy transfer in Bi/Yb-doped Gd2O3 phosphors, where they observed an increase in the NIR emission (980 nm) of Yb3+ ions when the sample was excited in the UV region.22a They have attributed the result to the transfer of Bi3+ 3P1 energy to two Yb3+ ions via the downconversion process. We emphasize that it is likely that the change in optical properties can be attributed to the change in the crystal phase, as has been observed in the TiO2 and NaYF4 matrices.25 However, we believe that with only a slight difference in energy levels between tetragonal and monoclinic BiVO4, it is hard to attribute the observed specific reduction in the blue emission intensity.
Based on the observed results, and from the above reports, we propose the following mechanism. The 1G4 level of the Tm3+ ions is populated via the Yb3+ ions via a multiphoton process, as shown in the inset of Fig. 3. This energy is subsequently transferred to the 3P1 level of the Bi3+ ions. From this level, the energy may decay to 3P0 and non-radiatively reach the ground state. Alternatively, the excited energy may also be transferred to the neighbouring Yb3+ ions. The latter might increase Yb3+ emission, but we could not measure it due to the detection limit of the spectrometer. The schematic representation of the proposed mechanism is given in Fig. 7.
 | ||
| Fig. 7 Schematic of the energy transfer mechanism involving Tm3+, Bi3+ and Yb3+ ions in Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 microcrystals. Dotted lines indicate non-radiative relaxations. | ||
To further verify the proposed energy transfer mechanism involving the Tm3+ to the Bi3+ 3P0 level, another control sample was prepared by doping 50% Gd3+ ions into YVO4 microcrystals in an identical experimental procedure. The reason for choosing Gd3+ is due to its close match in ionic size and charge to the Bi3+ ions and its lack of any energy level in the visible region that could match with the 1G4 level of the Tm3+ ions to alter the optical properties via energy transfer. The upconversion emission spectrum from the corresponding sample (shown in Fig. 8) exhibits strong blue emission centered at 475 nm, substantiating the claim that the observed reduction in intensity of the blue upconversion emission in Tm3+/Yb3+-doped Yi0.89−xBixVO4 microcrystals is caused by energy transfer from the 1G4 level of Tm3+ to the 3P0 levels of the Bi3+ ions.
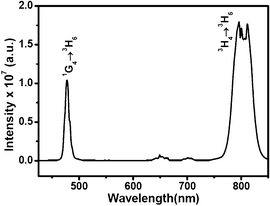 | ||
| Fig. 8 Upconversion emission spectrum of Tm3+(0.01)-doped Gd0.50Bi0.39VO4 microcrystals. | ||
Finally, to understand how the decrease in intensity of the blue emission with respect to other emissions can tune or enhance the colour mixing of the output light, we have calculated the CIE colour coordinates, which give the actual colour the human eye perceives. The colour coordinate values are calculated from the upconversion emission spectra of the Tm3+(0.01)/Yb3+(0.10)-doped Y0.89-xBixVO4 (x = 0 to 0.89) microcrystals. It is quite clear from the CIE diagram that with increasing Bi3+ concentration, the colour coordinates move from pure blue to pinkish blue (see Fig S8 of the ESI†). In fact for the sample Tm3+(0.01)/Yb3+(0.10)-doped BiVO4 microcrystals, the CIE coordinates fall almost close to the edge of the white light region.
Conclusion
In this paper we have reported that by controlling the Bi3+/Y3+ concentration in Tm3+(0.01)/Yb3+(0.10)-doped Y0.89−xBixVO4 (x = 0 to 0.89) microcrystals, it is feasible to tune the NIR/blue upconversion emission intensity of the Tm3+ ions. The blue emission intensity is preferentially reduced up to 800 times with respect to NIR emission by completely replacing the Y3+ ions with Bi3+ ions in Tm/Yb-doped Y0.89−xBixVO4 microcrystals. There is little doubt, from the control experiments, that Bi3+ plays a vital role in decreasing the intensity of the blue emission of the Tm3+ ions. An energy transfer mechanism involving Tm3+, Bi3+ and Yb3+ has been proposed for the preferential decrease in blue emission intensity. We strongly predict that the increase in the NIR/blue emission intensity, particularly obtained via upconversion, will be interesting in bio-imaging applications, as the penetration depth and consequently the contrast of the imaging process can be increased. Moreover, the preferential reduction in the blue emission intensity without altering the NIR emission near 800 nm results in a unique emission pattern, which can be utilized for anti-counterfeiting applications.Acknowledgements
MV would like to thank the Department of Science and Technology (DST) and Indian Institute of Science Education and Research (IISER), Kolkata for the funding. C. H. and S. S. thank the IISER- Kolkata and UGC, respectively for their fellowships. The authors thank Dr Sujatha Venkataraman at the University of Colorado at Denver and Dr Sri Sivakumar at IIT-Kanpur for helpful discussions. We thank Mr. Abhisek Basu for helping with Raman measurements.References
- (a) G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer, Berlin, 1994 Search PubMed; (b) A. Kitai, Luminescent Materials and Applications, John Wiley & Sons, West Sussex , 2008, Search PubMed; (c) C. R. Ronda, T. Jüstel and H. Nikol, J. Alloys Compd., 1998, 275–277, 669 CrossRef CAS; (d) S. Cotton, Lanthanide and Actinide Chemistry, 2006, John Wiley & Sons, West Sussex Search PubMed; (e) F. Vetrone, J.-C. Boyer and J. A. Capobianco, in The Handbook of Luminescence, Display Materials and Devices ed. H. S. Nalwa and L. S. Rohwer, American Scientific Publishers, 2003, vol. 3 Search PubMed; (f) T. Montini, A. Speghini, L. De Rogatis, B. Lorenzut, M. Bettinelli, M. Graziani and P. Fornasiero, J. Am. Chem. Soc., 2009, 131, 13155 CrossRef CAS.
- (a) A. J. Steckl, M. Garter, D. S. Lee, J. Heikenfeld and R. Birkhahn, Appl. Phys. Lett., 1999, 75, 2184 CrossRef CAS; (b) J. H. Kim and P. H. Holloway, Adv. Mater., 2005, 17, 91 Search PubMed; (c) A. de B.-Dias, Dalton Trans., 2007, 2229 RSC.
- (a) F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS; (b) S. Sivakumar, F. C. J. M. van Veggel and M. Raudsepp, J. Am. Chem. Soc., 2005, 127, 12464 CrossRef CAS; (c) S. Heer, O. Lehmann, M. Haase and H.-U. Güdel, Angew. Chem., Int. Ed., 2003, 42, 3179 CrossRef CAS; (d) M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef CAS; (e) F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS; (f) C. Li and J. Lin, J. Mater. Chem., 2010, 20, 6831 RSC.
- (a) V. Mahalingam, M. Francesca, F. Vetrone, V. Venkatramu, A. Speghini, M. Bettinelli and J. A. Capobianco, J. Phys. Chem. C, 2008, 112, 17745 CrossRef CAS; (b) J. Silver, E. Barrett, P. J. Marsh and R. J. Withnall, J. Phys. Chem. B, 2003, 107, 9236 Search PubMed; (c) F. Pandozzi, F. Vetrone, J.-C. Boyer, R. Naccache, J. A. Capobianco, A. Speghini and M. A. Bettinelli, J. Phys. Chem. B, 2005, 109, 17400 Search PubMed; (d) J. Silver, M. I. Martinez-Rubio, T. G. Ireland, G. R. Fern and R. Withnall, J. Phys. Chem. B, 2001, 105, 948 CrossRef CAS; (e) H. Schäfer, P. Ptacek, K. Kömpe and M. Haase, Chem. Mater., 2007, 19, 1396 CrossRef; (f) D. Chen, Y. Yu, F. Huang, P. Huang, A. Yang and Y. Wang, J. Am. Chem. Soc., 2010, 132, 9976 CrossRef CAS; (g) X. Zhang, P. Yang, C. Li, D. Wang, J. Xu, S. Gai and J. Lin, Chem. Commun., 2011, 47, 12143 RSC.
- (a) J. Lin, M. Yu, C. Lin and X. Liu, J. Phys. Chem. C, 2007, 111, 5835 CrossRef CAS; (b) V. Buissette, D. Giaume, T. Gacoin and J.-P. Boilot, J. Mater. Chem., 2006, 16, 529 RSC; (c) D. K. Williams, B. Bihari, B. M. Tissue and J. M. McHale, J. Phys. Chem. B, 1998, 102, 916 CrossRef CAS; (d) J. Liu and Y. Li, J. Mater. Chem., 2007, 17, 1797 RSC; (e) M. L. Pang, W. Y. Shen and J. Lin, J. Appl. Phys., 2005, 97, 033511 CrossRef.
- (a) L. Ozawa and M. Itoh, Chem. Rev., 2003, 103, 3835 CrossRef CAS; (b) F. Vetrone, J.-C. Boyer, J. A. Capobianco, A. Speghini and M. J. Bettinelli, J. Phys. Chem. B, 2003, 107, 10747 CrossRef CAS; (c) K. Riwotzki and M. J. Haase, J. Phys. Chem. B, 2001, 105, 12709 CrossRef CAS; (d) M. Yu, J. Lin, Z. Wang, J. Fu, S. Wang, H. J. Zhang and Y. C. Han, Chem. Mater., 2002, 14, 2224 CrossRef CAS.
- (a) H. H. Yang, H. Cheng, Y. G. Tang and Z. G. Lu, J. Am. Ceram. Soc., 2009, 92, 931 Search PubMed; (b) Y. Bai, Y. Wang, K. Yang, X. Zhang, G. Peng, Y. Song, Z. Pan and C. H. Wang, J. Phys. Chem. C, 2008, 112, 12259 CrossRef CAS; (c) M. L. Pang, W. Y. Shen and J. Lin, J. Appl. Phys., 2005, 97, 033511 CrossRef; (d) G. Chen, H. Liu, H. Liang, G. Somesfalean and Z. Zhang, J. Phys. Chem. C, 2008, 112, 12030 CrossRef CAS; (e) Z. Cheng, P. Ma, Z. Hou, W. Wang, Y. Dai, X. Zhai and J. Lin, Dalton Trans., 2012, 41, 1481 RSC; (f) P. Ghosh, K. Priolkar and A. Patra, J. Phys. Chem. C, 2007, 111, 571 CrossRef CAS; (g) S. Ray, A. Patra and P. Pramanik, Opt. Mater., 2007, 30, 608 Search PubMed; (h) A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, J. Phys. Chem. B, 2002, 106, 1909 CrossRef CAS; (i) G. S. Maciel, M. A. R. C. Alencar, C. B. de Araújo and A. Patra, J. Nanosci. Nanotechnol., 2010, 10, 2143 Search PubMed; (j) J.-C. Park, H.-K. Moon, D.-K. Kim, S.-H. Byeon, B.-C. Kim and K.-S. Suh, Appl. Phys. Lett., 2000, 77, 2162 CrossRef CAS; (k) G. Chen, H. Liu, H. Liang, G. Somesfalean and Z. Zhang, J. Phys. Chem. C, 2008, 112, 12030 CrossRef CAS.
- (a) W. J. Park, M. K. Jung, S. J. Im and D. H. Yoon, Colloids Surf., A, 2008, 313–314, 373 Search PubMed; (b) B. N. Mahalley, R. B. Pode and P. K. Gupta, Phys. Status Solidi A, 1999, 177, 293 Search PubMed.
- B. N. Mahalley, S. J. Dhoble, R.B. Pode and G. Alexander, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 39 CrossRef CAS.
- S. Yan, J. Zhang, X. Zhang, S. Lu, X. Ren, Z. Nie and X. J. Wang, J. Phys. Chem. C, 2007, 111, 13256 CrossRef CAS.
- C. Jacinto, M. V. D. Vermelho, E. A. Gouveia, M. T. de Araujo, P. T. Udo, N. G. C. Astrath and M. L. Baesso, Appl. Phys. Lett., 2007, 91, 071102 CrossRef.
- M. Nyk, R. Kumar, T. Y. Ohulchanskyy, E. J. Bergey and P. N. Prasad, Nano Lett., 2008, 8, 3834 CrossRef CAS.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839 RSC.
- V. Mahalingam, R. Naccache, F. Vetrone and J. A. Capobianco, Chem. Commun., 2011, 47, 3481 RSC.
- J. Ghamri, H. Baussart, M. Le and Brast and J.-M. Leroy, J. Phys. Chem. Solids, 1989, 50, 1237 CrossRef CAS.
- H. M. Zhang, J. B. Liu, H. Wang, W. X. Zhang and H. Yan, J. Nanopart. Res., 2008, 10, 767 CrossRef CAS.
- M. Gotić, S. Musić, M. Ivanda, M. Šoufek and S. J. Popović, J. Mol. Struct., 2005, 744–747, 535 CAS.
- J. B. Liu, H. Wang, S. Wang and H. Yan, Mater. Sci. Eng., B, 2003, 104, 36 CrossRef.
- B. G. Wybourne, Spectroscopic Properties of Rare Earths,Wiley, NewYork, 1965 Search PubMed.
- (a) G. Ju, Y. Hu, L. Chen, X. Wang, Z. Mu, H. Wu and F. J. Kang, J. Electrochem. Soc., 2011, 158, J294 CrossRef CAS; (b) G. Chen, T. Y. Ohulchanskyy, R. Kumar, H. Ågren and P. N. Prasad, ACS Nano, 2010, 4, 3163 CrossRef CAS; (c) S. Heer, O. Lehmann, M. Haase and H.-U. Güdel, Angew. Chem., Int. Ed., 2003, 42, 3179 CrossRef CAS.
- J. F Suyver, A. Aebischer, S. Garcia-Revilla, P. Gerner and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125123 CrossRef.
- (a) X. Y. Huang and Q. Y. Zhang, J. Appl. Phys., 2010, 107, 063505 Search PubMed; (b) G. Liu, Y. Zhang, J. Yin and W. Zhang, J. Lumin., 2008, 128, 2008 Search PubMed.
- A. A. Steler and A. M. Srivastava, Opt. Mater., 2006, 29, 410 CrossRef CAS.
- (a) X. Xiao and B. Yan, J. Alloys Compd., 2006, 421, 252 Search PubMed; (b) H.-D. Nguyen, S.-il. Mho and In.-H. Yeo, J. Lumin., 2009, 129, 1754 CrossRef CAS.
- (a) A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, Chem. Mater., 2003, 15, 3650 CrossRef CAS; (b) F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: SEM images, XRD and Raman spectra of Tm3+(0.01)-doped Yb0.89−xBixVO4 microcrystals and CIE color coordinate diagram. See DOI: 10.1039/c2ra20239e/ |
| This journal is © The Royal Society of Chemistry 2012 |