The first organocatalytic asymmetric synthesis of 3-substituted isoindolinones†
Vijaykumar
More
a,
Renate
Rohlmann
b,
Olga García
Mancheño
*b,
Carmen
Petronzi
a,
Laura
Palombi
a,
Antonio De
Rosa
a,
Antonia Di
Mola
c and
Antonio
Massa
*a
aDipartimento di Chimica e Biologia, Università di Salerno, Via Ponte Don Melillo 84084 – Fisciano, SA, Italy. E-mail: amassa@unisa.it; Fax: +39 089 96960; Tel: +39 089 969565
bInstitute of Organic Chemistry, Münster University, 48149, Münster, Germany
cDipartimento di Scienze Farmaceutiche, Università di Salerno, Via Ponte Don Melillo 84084 – Fisciano, SA, Italy
First published on 13th February 2012
Abstract
Herein we describe the first asymmetric organocatalytic synthesis of 3-substituted isoindolinones in a convenient aldol-cyclization-rearrangement tandem reaction of malonates with 2-cyanobenzaldehyde. Bifunctional thiourea-cinchona catalysts proved to be particularly effective, giving the title compounds in high yields and moderate to good enantiomeric excesses. Moreover an efficient process of reverse crystallization led to a further enrichment up to >99% ee.
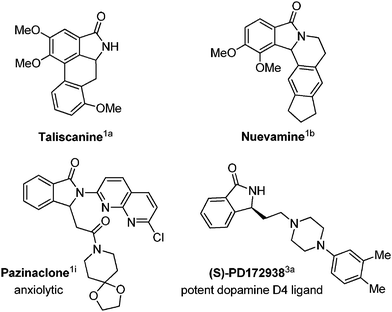
Isoindolinones are interesting heterocyclic compounds due to their presence in many naturally occurring substances,1a,b and for their large number of applications in therapeutic activities,1–4 like antihypertensive,1c antipsychotic,1d–f anesthetic,1g,h anxiolytic,1i antiviral,1j–l antileukemic.1m Some examples are described in Fig. 1. Therefore, great attention is devoted to the development of new methods for the synthesis of 3-substituted isoindolinones, which constitute valuable scaffolds in many synthetic routes.2–4
 | ||
| Fig. 1 Some bioactive substituted isoindolinones. | ||
However, in spite of the considerable interest in the field, the construction of this heterocyclic core often requires the use of metal catalysis and/or non-flexible multi-step synthesis.1–4 Conversely, Ramström et al. have recently described a convenient approach to nitro-alkyl-3-substitued isoindolinones exploiting a tandem Henry reaction-cyclization-rearrangement of 2-cyanobenzaldehyde catalysed by tertiary amines.5 Moreover, as part of our ongoing research on the challenging aldol additions of active methylene compounds,6 we have recently reported a more general approach to 3-substituted isoindolinones exploiting the aldol addition of several classes of readily enolizable 1,3-dicarbonyl compounds to 2-cyanobenzaldehyde in the presence of triethylamine.7 On the other hand, the asymmetric synthesis of 3-substituted isoindolinones remains challenging. Thus, though the resolution of racemic mixtures with chiral acids,3a bases3b or the use of chiral auxiliaries3c–e has been extensively exploited in the synthesis of enantioenriched isoindolinones, to the best of our knowledge only one catalytic asymmetric synthesis of 3-substituted isoindolinones has been reported.4 In this method Huang and coworkers used a chiral copper catalyst in a Michael addition/Mannich tandem reaction, leading to 3-substituted isoindolinones in good yields and enantioselectivities.4 Considering the obvious limitations of the current asymmetric synthesis of isoindolinones, the development of simple organocatalytic approaches are highly desirable.
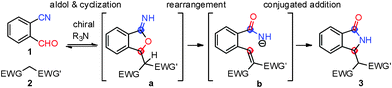
In order to tackle this challenge, we decided to explore the organocatalyzed asymmetric aldol-cyclization/rearrangement/Michael-type tandem reaction as shown in Scheme 1.
 | ||
| Scheme 1 Organocatalytic asymmetric approach to isoindolinones. | ||
Based on the accepted reaction mechanism for this transformation,5,7 we envisioned the use of bifunctional catalysts containing a tertiary amine and a thiourea moiety as a straightforward strategy via non-covalent activation.8–12 In fact, considering that the initial stereocentre formed during the aldol-type reaction is lost in the course of the reaction, to attain high levels of enantioselectivity, chiral thiourea as H–bond-donor for the activation of the last intramolecular conjugated addition of the tandem process could be crucial.
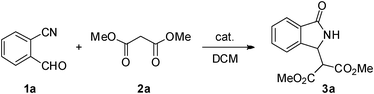
In order to verify the effectiveness of our hypothesis, we firstly focused our efforts on the model reaction between 2-cyano benzaldehyde (1a) and dimethyl malonate (2a) using quinine and other readily available bifunctional thiourea catalysts (Fig. 2).8–12
 | ||
| Fig. 2 Bifunctional thiourea catalysts. | ||
In a preliminary experiment, we observed that a catalytic amount (10 mol%) of quinine showed higher efficiency than the previously reported use of stochiometric amounts of triethylamine.7 In this way isoindolinone 3a was formed in an excellent 96% yield after 8 h at room temperature. However, as expected, a very low asymmetric induction was obtained (10% ee, Table 1, entry 1).13 On the other hand, when thiourea-cinchona catalyst 4, prepared from quinine in two steps,10 was employed under the same reaction conditions (10 mol% catalyst in DCM at r.t.), 3a was obtained in an excellent 98% yield and an encouraging enantioselectivity of 41% ee (entry 2). Decreasing the temperature to 0 °C, lower values of ee, as sometimes reported for thiourea-cinchona catalysts,9d were obtained (entry 3). On the other hand, a higher dilution (0.05 vs. 0.2 M) was beneficial, yielding 3a in a good 62% ee (entry 4). Under these conditions, other thiourea-cinchona catalysts 5, 6 and 7 were next tested. Similar results in terms of both reactivity and enantioselectivity were obtained with the related 6-demethoxy catalyst 6 (63% ee, entry 6). Conversely, their pseudo-enantiomers 5 and 7, led to the opposite enantiomer (ent-3a) in lower enantioselectivities (entries 5 and 7), showing the importance of the appropriately matched spatial placement of both the amine and thiourea units. Taking 4 as the optimal catalyst, an extensive screening of the reaction conditions was carried out. Thus, we could observe that a higher reaction temperature (50 °C oil bath or reflux vs. r.t.) gave an enhancement of the enantioselectivity (entry 8). On the other hand, we could decrease the amount of catalyst to 5 mol%, maintaining the same levels of enantioinduction (r.t. and reflux, entries 9 and 10). It is worthy to note that the good ee value of the last experiment was remarkably improved after only one efficient reverse crystallization process, providing isoindolinone 3a in good yield and a very high 99% ee (result in brackets, entry 10). Interestingly, from a preparative point of view, similar high enantioselectivities were obtained by crystallization of different enantioenriched samples of 3a and ent-3a, obtained with catalyst 4 and 5, respectively (up to 99% ee 3a and 96% ee ent-3a, entries 10 and 11). Under these conditions other bifunctional thiourea catalysts 8 and 9 were tested. In particular, the Takemoto catalyst 811 showed a comparable efficiency to 4, while 9 led to a remarkably lower enantioselectivity (entries 12 and 13).
| Entry | Cat. (mol%) | [M]b | t/h | T/°C | Yield (%)c | ee (%)d |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol) and catalyst in DCM. b [M] refers to 1a. c Isolated yield. d Determined by chiral HPLC (see ESI for details). e In brackets yield and ee after crystallization. | ||||||
| 1 | Quinine (10) | 0.2 | 8 | r.t. | 96 | 10 |
| 2 | 4 (10) | 0.2 | 3.5 | r.t | 98 | 41 |
| 3 | 4 (10) | 0.2 | 12 | 0 °C | 99 | 25 |
| 4 | 4 (10) | 0.05 | 24 | r.t. | 85 | 62 |
| 5 | 5 (10) | 0.05 | 24 | r.t. | 96 | −55 |
| 6 | 6 (10) | 0.05 | 28 | r.t. | 80 | 63 |
| 7 | 7 (10) | 0.05 | 24 | r.t. | 96 | −52 |
| 8 | 4 (10) | 0.05 | 24 | reflux | 96 | 66 |
| 9 | 4 (5) | 0.05 | 72 | r.t. | 92 | 62 |
| 10 | 4 (5) | 0.05 | 38 | reflux | 99 (56)e | 62 (>99)e |
| 11 | 5 (5) | 0.05 | 48 | reflux | 76 (23)e | −54 (−96)e |
| 12 | 8 (5) | 0.05 | 72 | r.t. | 95 (71)e | 63 (92)e |
| 13 | 9 (5) | 0.05 | 48 | reflux | 99 | −16 |
| 14 | 4 (15) | 0.017 | 72 | r.t. | 96 (67)e | 74 (96)e |
| 15 | 4 (15) | 0.017 | 48 | reflux | 81 (69)e | 70 (94)e |
| 16 | 4 (20) | 0.013 | 48 | reflux | 90 | 72 |
| 17 | 10 (15) | 0.017 | 48 | r.t. | 99 | 70 |
| 18 | 5 (15) | 0.017 | 72 | reflux | 82 (50)e | −64 (−94)e |
Thus, in order to achieve higher enantioselectivities, we reconsidered exploiting the positive trend observed by increasing the dilution. Due to the slower reaction rates under higher dilution and as a compromise between reactivity and enantioselectivity, 15 mol% of 4 in a 0.017 M solution of DCM at both room temperature and reflux was then employed (entries 14 and 15). As a result, the enantiomeric excess was improved up to 74%. Further dilution at 0.013 M in the presence of 20% mol of 4 led to the same level of ee (entry 16). Other solvents were also screened: DCM was confirmed to be the best, whereas THF was radically less effective and MeOH led to decomposition products. Under the 0.017 M dilution conditions, the cinchona-squarimide catalyst 1012 showed a similar efficiency to 4 (70% ee, entry 17). These conditions also provided an improvement of the ee of ent-3a in the presence of catalyst 5 (compare entry 18 with entry 11).
With these positive results in hand, the effect of other differently substituted malonates as nucleophiles in the presence of 15 and 5 mol% of 4 in DCM was next analysed (Table 2). Excellent yields and similar good enantiomeric excesses were obtained with diethylmalonate (entries 1 and 2). Interestingly, increasing the bulkiness of the substituent to an isopropyl group leads to the most efficient nucleophile. Thus, the corresponding isoindolinone 3c was obtained with a superior ee of 81% (entry 4). Conversely, di-tert-butyl-malonate was revealed to be less selective, probably due to its greater steric hindrance (entry 7). It is important to note that the latter nucleophile has proved totally ineffective in the non-asymmetric version in the presence of Et3N, emphasising the effectiveness of this methodology. On the other hand, a more flexible benzyl group on the malonate was less efficient. However, the process of reverse crystallization was again remarkably effective, permitting us to obtain a highly enantiopure isoindolinone 3e (up to >99% ee, entries 8 and 9). Finally, the reaction with dimethyl and di-isopropylmalonates was extended to other substituted 2-cyanobenzaldehyde derivatives (Fig. 3). For that purpose, we focused on three representative substrates with (i) a fluoride substituent (1f), (ii) two electron donating methoxy groups (1g), and (iii) a heterocyclic core such as a quinoline (1h). These substitution patterns are very attractive since they constitute important motifs for several biologically active compounds.1p–t,3c,3f
 | ||
| Fig. 3 Asymmetric synthesis of isoindolinones: substrate scope. | ||

| Entry | R | 4 (mol%) | t/h | Temp. | 3 | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol) and catalyst 4 (5 or 15 mol%) in DCM (0.05 M or 0.017 M, respectively). b Isolated yields. c Determined by chiral HPLC (see ESI for details). d Reaction with catalyst 10 gave 3c with the same 81% ee and 99% yield. e In brackets yield and ee after crystallization. | |||||||
| 1 | Et | 5 | 48 | reflux | 3b | 99 | 64 |
| 2 | Et | 15 | 48 | r.t. | 3b | 90 | 74 |
| 3 | i-Pr | 15 | 48 | reflux | 3c | 88 | 76 |
| 4 | i-Pr | 15 | 80 | r.t. | 3c | 90 | 81d |
| 5 | i-Pr | 5 | 72 | r.t. | 3c | 94 | 68 |
| 6 | i-Pr | 5 | 48 | reflux | 3c | 98 | 71 |
| 7 | t-Bu | 5 | 70 | reflux | 3d | 87 | 46 |
| 8 | Bn | 5 | 70 | r.t. | 3e | 95 (45)e | 55 (>99)e |
| 9 | Bn | 15 | 70 | r.t. | 3e | 85 (57)e | 60 (97)e |
Thus, we were pleased to observe that 4-fluoro-2-formylbenzonitrile (1f) was also a good substrate in the tandem process with dimethylmalonate in the presence of only 5 mol% of catalysts 4 and 5. Isoindolinones 3f and ent-3f were then obtained in high yields and moderate enantioselectivities. As expected, the use of di-isopropylmalonate led to 3i with a significantly improved 78% ee. The enantioselectivity obtained with 1f was comparable to 2-cyanobenzaldehyde (1a), while in the other two cases lower levels of asymmetric induction were observed. Moreover, the electron-rich aromatic compound 1g bearing two methoxy groups proved to be notably less reactive, leading to 3g in moderate yields. This effect was more dramatic in the case of the reaction with di-isopropylmalonate, requiring longer reaction times for a similar conversion.
The lower reactivity of 1g could be expected because of the effect of the electron-rich methoxy groups. Therefore this aldehyde is less reactive towards nucleophilic addition in the first step. Nevertheless, the reverse crystallization process was effective also for these isoindolinones and we easily achieved good ees (values in brackets, Fig. 3), pointing out that the combination of the organocatalysed asymmetric tandem reaction together with the crystallization is a straightforward way to get a wide range of highly enantiomeric enriched isoindolinones.
To account for our experimental results, in which simple chiral tertiary amines such as quinine or sparteine led to technical racemic compounds, whereas bifunctional thiourea catalysts such as thiourea-cinchona derivatives proved to be very efficient, we propose a possible cooperative interaction mode with these catalysts (Fig. 4). From a mechanistic point of view, focusing on the stereochemical determining step, these bifunctional catalysts can doubly activate intermediate bvia hydrogen-bonding interactions with the carboxylic groups and the amide moiety (see also Scheme 1). Thus, after the deprotonation and ring opening, an interaction between the quinuclidine moiety and the formed amide can be envisioned. This interaction would facilitate one facial approach of the internal N-nucleophile. At the same time, the activation of the Michael acceptor by H-bonding of the thiourea moiety with the carboxylic groups can be expected (Fig. 4).
 | ||
| Fig. 4 Possible cooperative action mode of bifuntional catalyst 4. | ||
In conclusion we have described the first asymmetric organo-catalytic synthesis of malonate-3-substituted isoindolinones exploiting an aldol addition/cyclization/rearrangement tandem process of a range of 2-cyanobenzaldehydes and malonates. Bifunctional thiourea-cinchona catalysts were revealed to be particularly effective and gave 3-substituted isoindolinones in high yields and moderate to good enatiomeric excesses. Moreover, an efficient and general process of reverse crystallization led to a further enrichment up to >99% ee. Considering the importance of 3-substituted isoindolinones in the synthesis of biologically active compounds and the simplicity of our method, further studies to improve the efficiency and to expand the scope to other classes of active methylene compounds and different substituted 2-cyanobenzaldehydes are in progress.
Acknowledgements
This work was supported by FARB funds of Italian Ministry of Education and by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie. R.R. thanks Münster University within the Bonusprogram for a predoctoral contract. See Supporting information for experimental procedures, NMR copies of new compounds and HPLC traces.References
- (a) For some naturally occurring substances see: Nuevamine: V. Fajardo, V. Elango, B. K. Cassels and M. Shamma, Tetrahedron Lett., 1982, 23, 39 CrossRef CAS; (b) Taliscanine: H. A. Priestap, Phytochemistry, 1985, 24, 848 CrossRef; (c) For the antihypertensive activity, see: J.-M. Ferland, C. A. Demerson and L. G. Humber, Can. J. Chem., 1985, 63, 361 CrossRef CAS; (d) For the antipsychotic activity, see: M. Linden, D. Hadler and S. Hofmann, Hum. Psychopharmacol., 1997, 12, 445 CrossRef CAS; (e) Z.-P. Zhuang, M.-P. Kung, M. Mu and H. F. Kung, J. Med. Chem., 1998, 41, 157 CrossRef CAS; (f) M. H. Norman, D. J. Minick and G. C. Rigdon, J. Med. Chem., 1996, 39, 149 CrossRef CAS; (g) For the anesthetic activity, see: Laboratori Baldacci, S. P. A. Japanese Patent 5,946,268, 1984; Chem. Abstr., 1984, 101, 54922 Search PubMed; (h) For the antiulcer activity, see: W. Lippmann, U.S. Patent4,267,189, 1981; Chem. Abstr., 1981, 95, 61988 Search PubMed; (i) For anxiolytic activity, see: Pazinaclone I. Takahashi, T. Kawakami, E. Hirano, H. Yokota and H. Kitajima, Synlett, 1996, 353 CrossRef CAS; (j) For the antiviral activity, see: T. R. Govindachari, K. R. Ravindranath and N. Viswanathan, J. Chem. Soc., Perkin Trans. 1, 1974, 1215 RSC; (k) I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623 CrossRef CAS; (l) E. De Clercq, J. Med. Chem., 1995, 38, 2491 CrossRef CAS; (m) For the antileukemic activity, see: E. C. Taylor, P. Zhou, L. D. Jenning, Z. Mao, B. Hu and J.-G. Jun, Tetrahedron Lett., 1997, 38, 521 CrossRef CAS; (n) For other interesting biological properties see also: H. P. Rang, M. M. Dale, J. M. Ritter, in Pharmacology, 3rd edn, Churchill-Livingstone, London, 1995 Search PubMed; (o) M. Anzini, A. Cappelli, S. Vomero, G. Giorgi, T. Langer, G. Bruni, M. R. Romeo and A. S. Basile, J. Med. Chem., 1996, 36, 4275 CrossRef; (p) M. Reiffen, W. Eberlein, P. Mueller, M. Psiorz, K. Noll, J. Heider, C. Lillie, W. Kobinger and P. Luger, J. Med. Chem., 1990, 33, 1496 CrossRef CAS; (q) L. Castedo, E. Guitian, J. M. Saa and R. Suau, Heterocycles, 1982, 19, 279 CrossRef CAS; (r) F. Pin, S. Comesse, M. Sanselme and A. J. Daiech, J. Org. Chem., 2008, 73, 1975 CrossRef CAS; (s) J. S. Yadav and B. V. S. Reddy, Tetrahedron Lett., 2002, 43, 1905 CrossRef CAS; (t) V. Machtey, H. E. Gottlieb and G. Byk, ARKIVOC, 2011, 9, 308 Search PubMed.
- Some recent examples of non-asymmetric metal catalysed isoindolinone formation: (a) C. Zhu and J. R. Falck, Org. Lett., 2011, 13, 1214 CrossRef CAS; (b) D. M. Shacklady-McAtee, S. Dasgupta and M. P. Watson, Org. Lett., 2011, 13, 3490 CrossRef CAS; (c) D. Augner, D. C. Gerbino, N. Slavov, J. M. Neudorfl and H.-G. Schmalz, Org. Lett., 2011, 13, 5374 CrossRef CAS; (d) D. Marosvolgyi-Hasko, A. Takacs, Z. Riedl and L. Kollar, Tetrahedron, 2011, 67, 1036 CrossRef; (e) D. D. Li, T. T. Yuan and G. W. Wang, Chem. Commun., 2011, 47, 12789 RSC; (f) J. W. Wrigglesworth, B. Cox, G. C. Lloyd-Jones and K. I. Booker-Milburn, Org. Lett., 2011, 13, 5326 CrossRef CAS.
- For other multi-step synthesis, see: (a) T. R. Belliotti, W. A. Brink, S. R. Kestern, J. R. Rubin, D. J. Wistrow, K. T. Zoski, S. Z. Whetzel, A. E. Corbin, T. A. Pugsley, T. G. Heffner and L. D. Wise, Bioorg. Med. Chem. Lett., 1998, 8, 1499 CrossRef CAS; (b) N. Kanamitsu, T. Osaki, Y. Itsuji, M. Yoshimura, H. Tsujimoto and M. Soga, Chem. Pharm. Bull., 2007, 55, 1682 CrossRef CAS; (c) D. Enders, V. Braig and G. Raabe, Can. J. Chem., 2001, 79, 1528 CrossRef CAS; (d) M. Lamblin, A. Couture, E. Deniau and P. Grandclaudon Tetrahedron, Tetrahedron: Asymmetry, 2008, 19, 111 CrossRef CAS; (e) S. Nieto, F. J. Sayago, P. Laborda, T. Soler, C. Cativiela and E. P. Urriolabeitia, Tetrahedron, 2011, 67, 4185 CrossRef CAS; (f) M. Lorion, A. Couture, E. Deniau and P. Grandclaudon, Synthesis, 2009, 1897 CAS.
- S. Guo, Y. Xie, X. Hu, C. Xia and H. Huang, Angew. Chem. Int. Ed., 2010, 49, 2728 CAS.
- (a) M. Angelin, M. Rahm, A. Fischer, T. Brinck and O. Ramström, J. Org. Chem., 2010, 75, 5882 CrossRef CAS; (b) M. Angelin, P. Vongvilai, A. Fischer and O. Ramström, Chem. Commun., 2008, 768 RSC; (c) M. Angelin, A. Fischer and O. Ramström, J. Org. Chem., 2008, 73, 3593 CrossRef CAS.
- (a) A. Massa, A. Scettri, R. Filosa and L. Capozzolo, Tetrahedron Lett., 2009, 50, 7318 CrossRef CAS; (b) A. Massa, A. Roscigno, P. De Caprariis, R. Filosa and A. Di Mola, Adv. Synth. Catal., 2010, 352, 3348 CrossRef CAS; (c) V. More, A. Di Mola, G. Croce, C. Tedesco, C. Petronzi, P. De Caprariis, A. Peduto, R. Filosa and A. Massa, Org. Biomol. Chem., 2011, 9, 8483 RSC.
- V. More, A. Di Mola, M. Perillo, P. De Caprariis, R. Filosa, A. Peduto and A. Massa, Synthesis, 2011, 3027 CAS.
- (a) P. M. Pihko, Hydrogen Bonding in Organic Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) C. E. Song, Cinchona Alkaloids in Synthesis and Catalysis, Wiley-VCH, Weinheim, 2009 Search PubMed; (c) S. J. Connon, Chem. Commun., 2008, 2499 RSC.
- For selected examples on conjugated addition reactions with bifunctional cinchona-thiourea catalysts, see: (a) T. Marcelli, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 7496 CrossRef CAS; (b) K. Liu, H.-F. Cui, J. Nie, K.-Y. Dong, X.-J. Li and J.-A. Ma, Org. Lett., 2007, 9, 923 CrossRef CAS; (c) N. J. A. Martin, L. Ozores and B. List, J. Am. Chem. Soc., 2007, 129, 8976 CrossRef CAS; (d) M. M. Biddle, M. Lin and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 3830 CrossRef CAS; (e) J. Alemán, A. Millelli, S. Cabrera, E. Reyes and K. A. Jørgensen, Chem.–Eur. J., 2008, 14, 10958 CrossRef; (f) X. Jiang, Y. Zhang, A. S. C. Chan and R. Wang, Org. Lett., 2009, 11, 153 CrossRef CAS; (g) O. García Mancheño, P. Tangen, R. Rohlmann, R. Fröhlich and J. Alemán, Chem.–Eur. J., 2011, 17, 984 CrossRef.
- For the synthesis of the bifunctional thiourea-cinchona catalysts, see: B. Vakulya, S. Varga, A. Csampai and T. Soos, Org. Lett., 2005, 7, 1967 CrossRef CAS.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS.
- For a recent review on squarimides in bifunctional organocatysis: J. Aleman, A. Parra, H. Jiang and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 6890 CrossRef CAS.
- No improvement of the enantioselectivity with quinine as catalyst was observed by changing the reaction conditions or using different alkaloids such as cupreidine, quinidine, cinchonine and cinchonidine (see supporting information for details).
Footnote |
| † Electronic supplementary information (ESI) available: The reaction times and conditions are detailed in tables. The equipments, the typical experimental procedures, spectroscopic data and HPLC traces are available in supporting information. See DOI: 10.1039/c2ra20231j |
| This journal is © The Royal Society of Chemistry 2012 |