Improvement of transport properties and hydrogen permeation of chemically-stable proton-conducting oxides based on the system BaZr1-x-yYxMyO3-δ†
Sonia
Escolástico
a,
Mariya
Ivanova
b,
Cecilia
Solís
a,
Stefan
Roitsch
c,
Wilhelm A.
Meulenberg
b and
José M.
Serra
*a
aInstituto de Tecnología Química (Universidad Politécnica de Valencia–Consejo Superior de Investigaciones Científicas), Av. Naranjos s/n, E-46022 Valencia, Spain. E-mail: jmserra@itq.upv.es; Fax: +34963877809
bForschungszentrum Jülich GmbH, Institute of Energy and Climate Research-IEK-1, Leo-Brandt-Str. 1, D-52425 Jülich, Germany
cErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons, RWTH Aachen University, Ahornstr. 55, 52074 Aachen, Germany
First published on 20th March 2012
Abstract
The structural and transport properties as well as the chemical stability of a series of proton-conducting oxides based on yttrium-doped barium zirconate were investigated. Specifically, Pr-, Fe- and Mn-doped BaZr1-x-yYxMyO3-δ compounds were prepared by solid state reaction. The compound exhibiting the highest total and protonic conductivity at elevated temperatures under reducing atmospheres was BaZr0.8Y0.15Mn0.05O3-δ. Temperature-programmed reduction experiments revealed a particular redox behavior related to the Mn-species under selected conditions. The hydrogen permeation was thoroughly studied as a function of the temperature, hydrogen concentration and the humidification degree in the sweep gas. Moreover, the transient processes induced by alternate step changes in the humidification degree of the sweep gas were analysed. The highest steady hydrogen evolution flow exceeded 0.03 ml min−1 cm−2 (0.9 mm-thick membrane) at 1000 °C for the humidified sweep gas. The stability of BaZr0.8Y0.15M0.05O3-δ under operation-relevant atmospheres (CO2-rich reducing atmosphere at high temperature) was tested using different techniques (X-ray diffraction (XRD), Raman, SEM, TEM and TG) and the results showed that this material is stable even when exposed to 115 ppm H2S.
1. Introduction
Hydrogen permeation membranes based on ceramic proton-conducting oxides1 may enable hydrogen purification and separation at high temperature, therefore making possible a high degree of integration in different industrial processes. Their implementation in water gas shift reactors in power plants applying fuel gasification schemes, e.g. IGCC, is of special importance. Process intensification due to thermodynamic displacement through in situ hydrogen extraction allows simplification of the process scheme and leads to an increase in the overall process efficiency and sustainability. Additionally, hydrogen separation enables CO2 sequestration in power plants operating on carbon-based fuels (fossil fuels or biomass-derived fuels). A prerequisite for membrane materials in this application is their stability at high temperatures in reducing and CO2-rich environments, along with sufficient hydrogen permeability.Many perovskite oxides, such as the doped BaCeO3, SrCeO3, SrZrO3 and BaZrO3 are reported to have reasonable proton conductivity in hydrogen-rich and humid atmospheres. From these groups, BaCeO3-based compounds exhibit the highest conductivity,2 however they suffer from insufficient chemical stability, especially in CO2-containing atmospheres. Exposure to CO2 leads to material degradation due to the formation of barium carbonate (BaCO3) and cerium oxide (CeO2).3 Nevertheless, BaZrO3-based compounds exhibit reasonable chemical stability but present lower conductivity, compared to the doped BaCeO3 compounds, due to higher grain boundary transport limitations.4,5 Moreover, the preparation of membranes made of doped BaZrO3 requires high sintering temperatures to achieve appropriate densities or gas tightness, although different approaches have been proposed to circumvent this problem.5–7 In order to achieve a compromise between proton conductivity and chemical stability, mixed solid solutions of BaCeO3 and BaZrO3 were developed, as reported elsewhere.8,9
In order to be implemented as hydrogen separation membranes, these materials ought to present mixed proton–electron conductivity with a stable proton conductivity term and a sufficient electronic conductivity term. The first strategy is to develop composite membranes comprising a mixture of the proton-conducting ceramic and another purely electronic conducting material to enhance the electronic conduction.10 The most promising approach involves a material tailored to boost the mixed conductivity, as reported in ref. 9, where electronic transport may be enhanced by means of intrinsic doping of the materials with multivalent cations. Concretely, BaCeO3 was doped with Eu and Sm by Wachsman et al., and as a result the ambipolar conductivity and hydrogen permeation were improved.11–13 Other important works have explored the doping of (Ba,Sr)CeO3 with Tb,14 Tm,15 Yb16 and Nd.17 A recent study18 on thin supported membranes manufactured from Eu-doped Sr(Zr,Ce)O3 solid solutions, reports peak hydrogen flow exceeding 0.5 ml min−1 cm−2 at 900 °C for a 20 μm-thick membrane. Recently, systematic studies19–21 on barium zirconate have illustrated the high potential of these materials for practical application as hydrogen membranes and ceramic proton-conducting fuel cell components.
In this work, the effect of B-site partial substitution in the perovskite-structured BaZr0.9Y0.1O3-δ material on ionic and electronic conductivity was studied. As a reference compound, the state-of-the-art protonic conductor BaZr0.9Y0.1O3-δ was selected due to its high bulk protonic conductivity,22,23 although the conductivity in polycrystalline samples drops due to the high grain boundary resistance reported for this material.24–27 Since hydrogen permeation requires mixed proton–electron conductivity, the structure and electrical properties of the reference material were modified by incorporating different cations (Fe, Mn and Pr) with variable oxidation states on the B-site position in the perovskite, aiming to improve the electronic conductivity. Moreover, the incorporation of these elements could also affect the grain boundary resistance by decreasing it. The present contribution shows a structural study of these compounds, their electrical conductivity under operating conditions, their stability under application-oriented atmospheres and the H2-permeation of a selected compound.
2. Experimental
2.1. Sample preparation
The solid state reaction method was used for the manufacturing of the compounds in the series BaZr1-x-yYxMyO3-δ studied in this work. For this purpose, BaCO3, ZrO2, Y2O3 and the corresponding metal oxides were weighed in stoichiometric proportions, thoroughly mixed and ball milled for 10 h. Subsequent calcination followed at 1400 °C for 32 h, and the resulting materials were ball milled for 1 day using zirconia balls.Rectangular bar samples used in the electrochemical study were prepared from the resulting BaZr1-x-yYxMyO3-δ (where x = 0.1 when y = 0, 0.05 and x = 0.15 when y = 0.05) powders via uniaxial pressing at 100 MPa. The green geometry of the bars was 40 × 5 × 4 mm3. The samples were sintered in air at 1700 °C for 30 h, obtaining a density of about 99%, as determined by the Archimedes method.
For the permeation tests in the present work, disc shaped samples with a diameter of 15 mm (in the sintered state) were uniaxially pressed at 72 MPa and sintered at 1700 °C for 30 h in air. Both disc sides were coated by screen printing with a 20 μm layer of Pt ink (Mateck, Germany), aiming to improve surface catalytic behavior. Pt coatings are not connected on the side.
2.2. Characterisation techniques
The microstructure of the sintered samples was characterized by means of scanning electron microscope Zeiss Ultra55 and transmission electron microscope FEI Tecnai F20 (with an acceleration voltage of 200 kV). TEM lamellas were prepared by means of a FIB (Focused-Ion Beam, FEI Helios Nanolab 400 s), with the subsequent thinning performed in a BAL-TEC RES-120.
Furthermore, the hydration and isotopic effects were studied in: (1) dry helium and helium saturated with H2O and D2O; (2) dry oxygen and moist oxygen with H2O and D2O; (3) dry hydrogen, hydrogen saturated with H2O, dry deuterium and deuterium saturated with D2O. In all experiments using H2O and D2O, the saturation was carried out at room temperature.
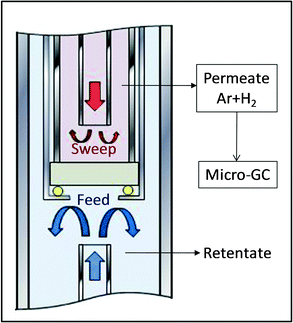 | ||
| Fig. 1 Schematic illustration of the permeation rig used, having two chambers separated by a gold-sealed dense ceramic membrane. | ||
(1) Treatment in a continuous gas flow of 10% CO2 and 90% CH4, both dry and humidified (2.5% water) at 700 °C and 800 °C for 72 h.
(2) Treatment under continuous gas flow composed of 115 ppm H2S, 4.43% CO2, 2.12% CO and 92.09% H2 at 500 °C, 30 bars, for 40 h.
The stability was controlled by XRD analysis before and after the treatments in order to observe microstructural and/or phase changes in the materials as a result of this short-term reduction of environmental exposure.
In addition, thermogravimetric (TG) measurements were performed on powder samples exposed to dry CO2 (5% in Ar) flow in the temperature range from 25 °C to 1000 °C with a heating ramp of 10 K min−1. For this purpose a Metler-Toledo StarE balance was used.
In order to check the in situ changes of the powders, Raman spectra were gathered using a CCR1000 Linkan Raman stage. The BaZr1-x-yYxMyO3 powders were heated up to 850 °C in oxygen, then the atmosphere was changed to 15% CO2 in Ar and then cooled down to 25 °C without changing the gas environment. Raman analysis was performed with a Renishaw inVia Raman spectrometer equipped with a Leica DMLM microscope and a 514 nm Ar+ ion laser as an excitation source. A ×50 objective of 8 mm optical length was used to focus the depolarized laser beam on a spot of 3 μm in diameter. A CCD array detector was used for collection of the Raman scattering.
3. Results and discussion
BaZr0.9Y0.1O3-δ was doped with Pr, Fe and Mn, aiming to modify the initial structure and create chemical defects, as well as to test the electrochemical and transport properties of the parent compound. Furthermore, in the Mn-doped compound the quantity of Y was also increased, aiming to improve specifically the protonic conductivity. For these reasons the selected compositions for this study were: BaZr0.9Y0.1O3-δ, BaZr0.85Y0.1Pr0.05O3-δ, BaZr0.85Y0.1Fe0.05O3-δ, BaZr0.85Y0.1Mn0.05O3-δ and BaZr0.8Y0.15Mn0.05O3-δ.3.1. Structural and microstructural characterization
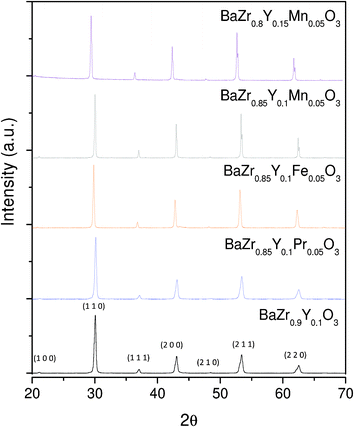 | ||
| Fig. 2 XRD patterns of the BaZr0.9Y0.1O3-δ, BaZr0.85Y0.1Pr0.05O3-δ, BaZr0.85Y0.1Fe0.05O3-δ,, BaZr0.85Y0.1Mn0.05O3-δ, and BaZr0.8Y0.15Mn0.05O3-δ, as sintered at 1400 °C. | ||
| Compound | Sample name | Lattice parameter a/Å at RT |
|---|---|---|
| BaZrO328 | BZ | 4.1913 |
| BaZr0.95Y0.1O3-δ | BZY | 4.1968 |
| BaZr0.85Y0.1Pr0.05O3-δ | BZYP | 4.1993 |
| BaZr0.85Y0.1Fe0.05O3-δ | BZYF | 4.2162 |
| BaZr0.85Y0.1Mn0.05O3-δ | BZYM1 | 4.2048 |
| BaZr0.8Y0.15Mn0.05O3-δ | BZYM2 | 4.2185 |
3.2. Electrochemical characterization
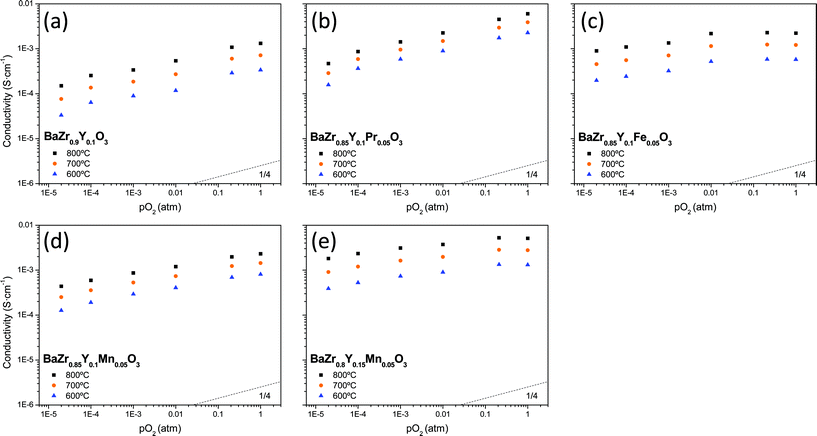 | ||
| Fig. 3 Total conductivity as a function of pO2 measured for five different compounds under dry conditions: (a) BaZr0.9Y0.1O3-δ, (b) BaZr0.85Y0.1Pr0.05O3-δ, (c) BaZr0.85Y0.1Fe0.05O3-δ, (d) BaZr0.85Y0.1Mn0.05O3-δ and (e) BaZr0.8Y0.15Mn0.05O3-δ. | ||
| Defect chemistry equations | ||
|---|---|---|
| Oxo↔v··o + O′′I | K F (T) = [v··o] × [O′′I] | (1) |
| nill ↔e′ + h· | Ke(T) = n × p | (2) |
| Oxo ↔ 1/2 O2(g) + v··o + 2e′ | K R (T) = [v··o] × n2 × pO21/2 | (3) |
Let us consider the compound BaZr0.9Y0.1O3-δ, called reference in the present study, in order to introduce the defect model. When Zr is partially replaced by a trivalent cation, such as Y3+, the oxygen vacancies are concurrently generated to compensate for the negative acceptor charge introduced by the incorporation of Y3+ in the Zr4+ position, as it is expressed in eqn (4):
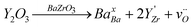 | (4) |
The electroneutrality condition can be then expressed by eqn (5).
| [Y′Zr] + 2[O′′i] + n = 2[v··o] + p | (5) |
By substituting into the defect equations (eqn (4)), in the high pO2 range, it is found that the concentration of electron holes is proportional to pO2 with a slope of ¼, e.g. p ∝ pO21/4. The ionic conductivity is, furthermore, independent of pO2, in accordance with the term (σi ∝ pO02).
With respect to the incorporation of a multivalent dopant such as Pr, the oxidation state changes (e.g. Pr3+ and Pr4+) depending on pO2 and the temperature. Subsequently, two different cases can be distinguished: (1) when Pr exists as Pr4+ (extreme oxidizing conditions), it does not contribute to the formation of new defects in the BaZr0.9Y0.1O3-δ parent compound; (2) when Pr exists as Pr3+, it generates oxygen vacancies following the same defect chemistry as in the case of Y-doped BaZrO3. The total nominal concentration of the formed charge-compensating defects is then higher than in the single acceptor dopant (as Y) initially used in the reference compound. Due to the reversibility of the oxidation state of these cations under given environmental conditions, charge compensation can also take place by the generation or annihilation of electrons or electrons holes, which results in substantial changes in electronic conductivity. As a consequence, the electroneutrality conditions may be formulated accordingly (eqn 6):
| [Y′Zr] + 2[O′′i] + [Pr′Zr] + n = 2[v··o] + p | (6) |
In the same way it is found that the concentration of electron holes is proportional to pO2 with a slope of ¼.
Another issue is dopant trapping, which is usually more pronounced when multi-dopant schemes or higher doping concentrations are used. Moreover, in multi-dopant compounds, acceptor doping effects (Y) and donor doping effects (Pr) may compensate depending on dopant concentration and environmental conditions.
In the case of Fe and Mn cations used as dopants in the referent BaZr0.9Y0.1O3-δ compound, a slightly different defect chemistry situation can be considered as they can also be in the +2 oxidation state, along with the previously discussed +3 and +4 states. In this case, the electroneutrality condition can be expressed by eqn (7) and eqn (8) for Fe and Mn, respectively.
| [Y′Zr] + 2[O′′i] + [Fe′Zr] + [Fe′′Zr] + n = 2[v··o] + p | (7) |
| [Y′Zr] + 2[O′′i] + [Mn′Zr] + 2[Mn′′Zr] + n = 2[v··o] + p | (8) |
As has been mentioned above, Fe and Mn doped compounds exhibit conductivity that is slightly less dependent on the pO2 than those having a slope of ¼ (Fig. 3). This effect can be ascribed to a transition situation between the two limiting defect states, the pure p-type electronic regime (with pO21/4 dependency) and the pure ionic regime (independent of pO2).
Fig. 4 depicts total conductivity against inverse temperature in dry O2, O2 + 2.5% H2O and O2 + 2.5% D2O for the five studied compounds. Two different trends in the behavior can be observed depending on the temperature range:
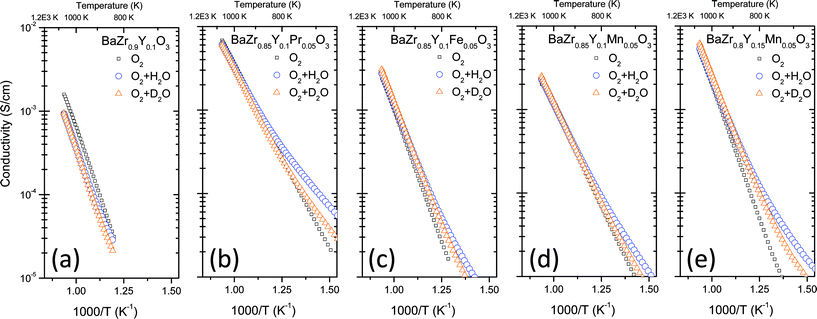 | ||
| Fig. 4 Total conductivity as a function of inverse temperature measured in dry oxygen, oxygen saturated with water and oxygen saturated with deuterated water: (a) BaZr0.9Y0.1O3-δ, (b) BaZr0.85Y0.1Pr0.05O3-δ, (c) BaZr0.85Y0.1Fe0.05O3-δ, (d) BaZr0.85Y0.1Mn0.05O3-δ and (e) BaZr0.8Y0.15Mn0.05O3-δ. Saturation with H2O and D2O was carried out at room temperature. | ||
1. Predominant protonic conductivity at low temperatures: the hydration effect  and the isotopic effect
and the isotopic effect  are evident for all tested compounds below 600 °C.
are evident for all tested compounds below 600 °C.
2. Predominant p-type and oxygen-ion conductivity at high temperatures: in these conditions the hydration and isotopic effects become negligible. This behavior is evident in the five tested compounds at temperatures above 600 °C due to oxide dehydration.
The same trends were observed for the measurements carried out in dry and wet helium (Fig. S1, ESI†). Furthermore, conductivity values in dry reducing (5% H2 and 5% D2) and wet reducing (5% H2 + 2.5% H2O and 5% D2 + 2.5% D2O) atmospheres are shown in Fig. 5 as a function of temperature. These conditions are the most relevant for practical application as H2-permeable membranes. It can be noticed that, in all studied compounds, protonic conduction prevails across the whole range of temperatures. This behavior can be inferred from the hydration effect ( ) and the isotopic effect (
) and the isotopic effect (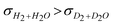 ) observed for all tested compounds.
) observed for all tested compounds.
 | ||
| Fig. 5 Total conductivity as a function of inverse temperature measured in hydrogen saturated with water and deuterium saturated with deuterated water for five different compounds: (a) BaZr0.9Y0.1O3-δ, (b) BaZr0.85Y0.1Pr0.05O3-δ, (c) BaZr0.85Y0.1Fe0.05O3-δ, (d) BaZr0.85Y0.1Mn0.05O3-δ and (e) BaZr0.8Y0.15Mn0.05O3-δ. Saturation with H2O and D2O was carried out at room temperature. | ||
At the highest temperature tested, the change in activation energy is related to the depletion of protons in the oxide and, as a consequence, the contribution of electrons and especially oxide ions becomes more relevant with the increase in temperature. Thus, the activation energy for proton and deuterium diffusion can be determined from the low temperature range (up to 600 °C) of the conductivity plots. Table 3 summarizes the activation energy values of the different compounds measured in oxidizing conditions. The very high values of these activation energies can be related to the grain boundary reported values,25,26 due to the limiting grain boundary resistivity for proton transport of this kind of material, deduced from the reported impedance studies.24–26 Furthermore, the activation energy for deuterium diffusion is about 0.04–0.08 eV higher than for protons (except for the Pr doped compound, which is higher). This difference is expected due to the lower zero point energy for deuterium ions than for protons.30
| Compound | O2+H2O Ea (eV) | O2+D2O Ea (eV) |
|---|---|---|
| BaZr0.95Y0.1O3-δ | 1.35 | 1.44 |
| BaZr0.85Y0.1Pr0.05O3-δ | 0.79 | 1.02 |
| BaZr0.85Y0.1Fe0.05O3-δ | 0.68 | 0.75 |
| BaZr0.85Y0.1Mn0.05O3-δ | 0.92 | 1.01 |
| BaZr0.8Y0.15Mn0.05O3-δ | 0.92 | 0.97 |
We should also emphasize the important improvement in total conductivity with increase in Y concentration, i.e. the change from BaZr0.85Y0.1Mn0.05O3-δ to BaZr0.8Y0.15Mn0.05O3-δ. Indeed, impedance analysis of BaZr0.8Y0.15Mn0.05O3-δ at 300 °C in dry and wet hydrogen (4% in Ar) revealed that the most important contribution to the total sample resistance is the one related to grain boundary transport (Fig. S2, ESI†). Moreover, it should be noted that our BaZr0.9Y0.1O3-δ reference compound shows total conductivity values lower than those reported elsewhere24–26 and this might be ascribed to (i) barium evaporation, (ii) non-stoichiometry, (iii) core-space charge layer effects, etc.25 Nevertheless, the best material found here (BaZr0.8Y0.15Mn0.05O3-δ) exhibits a conductivity almost two orders of magnitude higher than our reference compound (BaZr0.9Y0.1O3-δ). Furthermore, the protonic conductivity is enhanced in the Y-doped compound, as can be deduced from the higher magnitude of the isotopic effect in BaZr0.8Y0.15Mn0.05O3-δ than in BaZr0.85Y0.1Mn0.05O3. This fact is in agreement with previously reported studies31 and can be directly ascribed to the higher density of oxygen vacancies created by the substitution of Zr by Y, essential for the incorporation of protons in the oxide. As the protonic conductivity in these polycrystalline samples is principally limited by grain boundary resistance,24–26 the observed conductivity improvements should be a consequence of better grain boundary transport of both ions and electronic charge carriers achieved through the variation in composition.
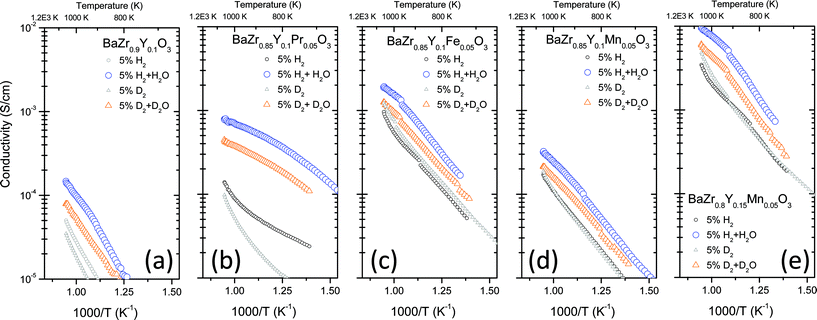
Aiming to understand the evolution of the Mn-oxidation state in BaZr0.8Y0.15Mn0.05O3-δ under reducing conditions, a temperature-programmed reduction (TPR) experiment was carried out in dry hydrogen. Fig. 6 presents the weight-normalized hydrogen consumption for BaZr0.9Y0.1O3-δ and BaZr0.8Y0.15Mn0.05O3-δ. The reference samples presents some background reduction in the temperature range 450 to 900 °C, which can be ascribed to oxygen release and the formation of electron carriers upon oxide dehydration. In the case of BaZr0.8Y0.15Mn0.05O3-δ, two additional pronounced reduction peaks are observed. The low temperature peak detected at 415 °C is related to the reduction of Mn4+ to Mn3+ and the consequent formation of the oxygen vacancies and water, which is in agreement with previous studies on La1-xCaxMnO3-δ perovskites.32,33 The reduction process taking place at 788 °C is related to the subsequent reduction of Mn3+ to Mn2+ and consumes a larger amount of hydrogen than the low temperature reduction. From the hydrogen consumption values, it can be inferred that Mn2+-ions are the prevailing species at temperatures above 900 °C in dry hydrogen while Mn4+-ions represent around 30% of the lattice Mn in perovskite at temperatures below 350 °C. Under the usual membrane operating conditions (750–1000 °C), Mn cations can be easily reduced or oxidized, depending on the specific temperature, pH2 and pH2O, which in turn can promote both the n-type electronic conduction and redox surface catalysis.
 | ||
| Fig. 6 Temperature-programmed reduction experiment in dry H2 (10% in Ar) for BaZr0.9Y0.1O3-δ and BaZr0.8Y0.15Mn0.05O3-δ compounds. | ||
3.3. H2 permeation study
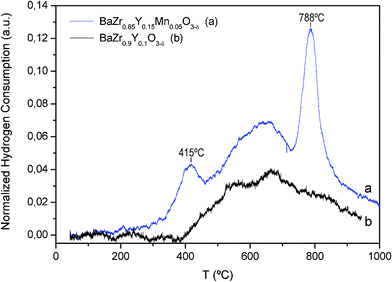
Hydrogen flux through the BaZr0.8Y0.15Mn0.05O3-δ membrane was studied as a function of several parameters as considered below.Fig. 7a shows hydrogen permeation fluxes of both configurations (hydration degree configurations) as a function of temperature, using 50% H2 as the feed gas. A clear improvement in hydrogen flux through the membrane can be observed when both sides are humidified due to (1) better hydration of the membrane, which results in a higher proton concentration across the membrane, especially in the region close to the permeate; and (2) the positive contribution of the oxide-ion transport in the direction of the feed, which leads to the formation of hydrogen in the permeate side, thanks to the water splitting process. Indeed, the pO2 gradient across the membrane and the associated oxide-ion transport direction are reversed when sweep gas is humidified. Therefore, when the sweep is dry (C1), part of the protons diffusing towards the permeate recombine with oxide-ions to form water and the net hydrogen flow is diminished. A similar behavior was previously observed in other hydrogen-permeable ceramic membranes.34
 | ||
| Fig. 7 Hydrogen flux through the BaZr0.8Y0.15Mn0.05O3-δ membrane: (a) flux as a function of temperature in two configurations: (i) when the feed side was humidified and (ii) when both feed and sweep sides of the membrane were humidified (the measurements were performed with a mixture of 50% H2 in He as the feed gas); (b) time stabilization of hydrogen flux at 1000 °C when the sweep gas is switched from wet to dry argon; and (c) flux at 1000 °C as a function of time, with several transitions from wet to dry argon sweep and vice versa. (Stages where water was added or removed are additionally notified by arrows.) | ||
Fig. 7b represents the transient process recorded at 1000 °C when switching from configuration C1 to C2. In the initial configuration with the dry sweep gas (C1), the hydrogen flux is lower than 0.01 ml min−1 cm−2 and the hydrogen flow increases steeply before reaching a peak value of 0.8 ml min−1 cm−2 upon switching to humidified sweep gas (C2). Nevertheless, the flow decreases drastically with time and after ∼100–150 min it stabilizes at around 0.03 ml min−1 cm−2, as can be seen in Fig. 7b. This temporary increase in hydrogen flux cannot be only attributed to the aforementioned higher membrane degree of hydration and simple water splitting effects on the sweep side. A plausible explanation is the potential partial oxidation of the membrane material when pO2 is increased on the sweep side, as a consequence of the addition of water. This oxidation could originate from the redox activity of Mn cations in this temperature range, involving the oxidation from Mn2+ to Mn3+. Therefore, it may be suggested that the initially observed increase in H2 flow to peak values of ∼0.8 ml min−1 cm−2 stems from coupled water splitting and membrane oxidation processes. Hence, the transient profile may be related to oxide-ion diffusion into the membrane. Moreover, in the steady state (C2), a graded concentration of Mn2+ and Mn3+ across the membrane would be expected from the applied pO2 gradient. Finally, this mechanism suggests that the humidification of the sweep induces several changes in the membrane material, that is, it alters the hydration degree, the concentration of oxygen vacancies and the concentration of electron carriers (n).
The study of the flux evolution at 1000 °C when the hydration configuration is successively changed (C1↔C2) is represented in Fig. 7c (note the logarithmic scale). Three different steps are plotted: (1) when both sides are humidified (C1→C2), the hydrogen flow passes through a maximum and then with time stabilizes around 0.03 ml min−1 cm−2; (2) when the sweep side is dried (C2→C1), the flux drops drastically, but later it increases slowly before reaching values lower than 0.01 ml min−1 cm−2 and finally (3) when the sweep side is humidified again (C1→C2), hydrogen flux increases quickly reaching a maximum and then the flow stabilizes at the same value as in the first step, around 0.03 ml min−1 cm−2.
The steep drop in hydrogen flow is consistent with the proposed transient mechanism and in this step change the membrane is reduced again, i.e. Mn3+ to Mn2+, consuming part of the hydrogen permeated during this temporary process. Moreover, the possible interpretation of the observed drastic drop in H2 flow due to the eventual degradation effects under the test conditions is ruled out, since this drop could be repeated in successive gas environment step changes and because of the reversibility of the process. Additionally, the results of the SEM and TEM studies support the membrane stability after the permeation test and allow the rejection such a possibility for explaining the observed flux degradation (see SEM and TEM images in Fig. 9 and 10, respectively).
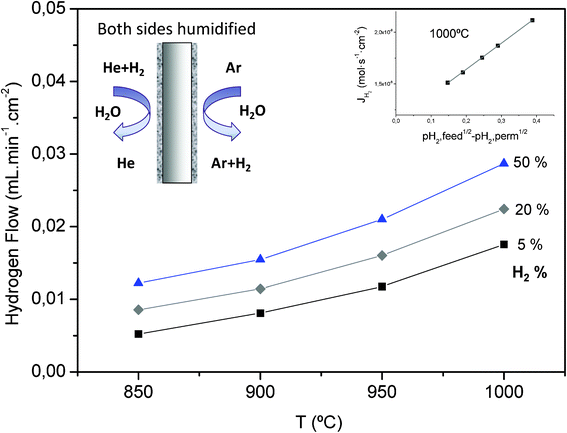 | ||
| Fig. 8 Hydrogen flux through the BaZr0.8Y0.15Mn0.05O3-δ membrane as a function of temperature and hydrogen partial pressure when both sides (feed and sweep) of the membrane are humidified. | ||
Based on the permeation measurements carried out in this work, and also considering Wagner theory, a flux of 7 ml min−1 cm−2 at 900 °C may be predicted for 20 μm-thick BaZr0.8Y0.15M0.05O3-δ membranes. When compared to the H2-flux measured for selected state-of-the art materials, e.g. 0.102 ml min−1 cm−2 for a 50 μm-thick SrCe0.95Y0.05O3-δ membrane at 950 °C (80% H2–He upstream and Ar downstream)36,37 or 13.44 ml min−1 cm−2 for a 2 μm-thick SrCe0.95Yb0.05O3-δ membrane at 677 °C (H2 upstream and He downstream),38 the predicted value seems to be very promising. Therefore, BaZr0.8Y0.15M0.05O3-δ can be pointed out as an attractive membrane material for H2-separation at elevated temperatures.
After hydrogen permeation measurements, the sample was analyzed by SEM, in order to check the stability of these compounds in humidified reducing conditions. Fig. 9 shows SEM images of the polished cross-section BaZr0.8Y0.15Mn0.05O3-δ membrane after the conductivity test, and no degradation or secondary phases in the membrane interior were detected. The average grain size is around 1 μm for the fresh and treated samples while the grain size for the BaZr0.85Y0.1Mn0.05O3-δ compound is slightly higher, around 2 μm. All investigated samples presented a very high density with very good attachment between grains and only small and scarce occluded pores can be found.
 | ||
| Fig. 9 SEM images of polished cross-sections of the BaZr0.8Y0.15Mn0.05O3-δ membrane before and after the hydrogen permeation test. | ||
Fig. 10 presents a transmission electron microscopy (TEM) study of the membrane region close to the surface exposed to the sweep side during the permeation test. Fig. 10c shows a micrograph of the cross section of the sample. Grains of 400 nm to 2 μm in diameter are present within the specimen. In Fig. 10a a sample area with larger magnification is visible, which lies approximately 2 μm below the sample surface. Neither secondary phases nor precipitates can be observed within the grains or at the grain boundaries, which confirms the total solution of the Y and Mn dopants, as suggested by the previous XRD analysis. Fig. 10b shows the sample at the specimen surface. The inhomogeneous layer at the top of the image corresponds to the residue of a platinum covering layer, which was deposited onto the sample during the specimen-preparation procedure. Between the covering layer and the sample a thin carbon layer (10–20 nm in thickness) with bright contrast is visible. Additionally, an enhanced surface roughness can be observed, which is considered to be a result of the permeation measurements. In line with this observation, Fig. 10d shows an SEM image of the top surface of the membrane. Grain boundaries cannot be distinguished in this micrograph, due to the surface roughness and the carbon layer. Occasionally, particles are observed on top of the surface of the membrane, which may originate from deposition of dust or particulate from piping and upstream setup components. This kind of surface foiling was previously reported for high temperature membranes tested in a similar experimental setup.39

3.4. Stability in reducing and CO2-rich environments
Fig. 11 shows the XRD patterns of the zirconate samples developed in this study and a reference sample made of Eu-doped BaCeO3. The short-term stability test was carried out on samples previously calcined at 1400 °C for 8 h. The powder samples were exposed to a continuous gas flow composed of 10% CO2 and 90% CH4 at 800 °C for 72 h. These conditions were selected in order to simulate the relevant hydrogen separation conditions, i.e. moist CO2-rich reducing atmospheres at elevated temperature. The temperature of 800 °C is high enough to test the samples against carbonate formation but still below the limit of carbonate decomposition. As seen from the figure, all tested zirconate samples remained apparently unchanged after treatment under the test conditions, while the Eu-doped BaCeO3 sample fully decomposed due to the formation of barium carbonates.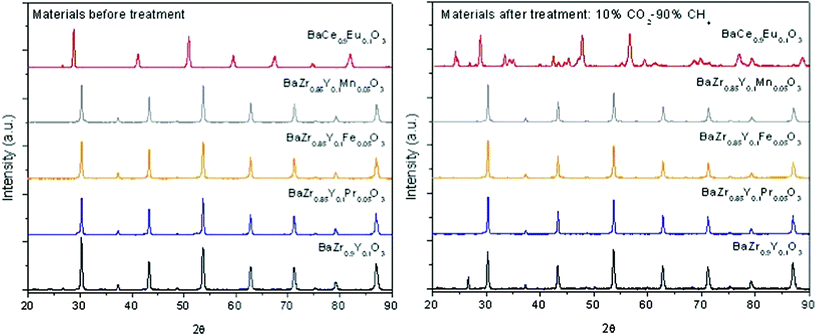 | ||
| Fig. 11 XRD diffraction patterns of compounds from the BaZr1-x-yYxMyO3-δ-series before and after the stability test in a mixture of 10% CO2 and 90% CH4 carried out at 800 °C for 72 h. The XRD diffraction pattern of BaCe0.9Eu0.1O3-δ was added for comparison. | ||
In a second stability test, the materials developed in this study were exposed for 40 h under the action of a continuous gas flow composed of 115 ppm H2S, 4.43% CO2, 2.12% CO and 92.09% H2 at 500 °C, 30 bars and the phase composition was controlled via the XRD. Pre- and post-treatment XRD-patterns are presented in Fig. 12. No secondary phases can be observed in the case of zirconates also for this set of experimental conditions. Nevertheless, the Eu-doped BaCeO3 decomposed again, forming barium carbonates and cerium-rich sulfates.
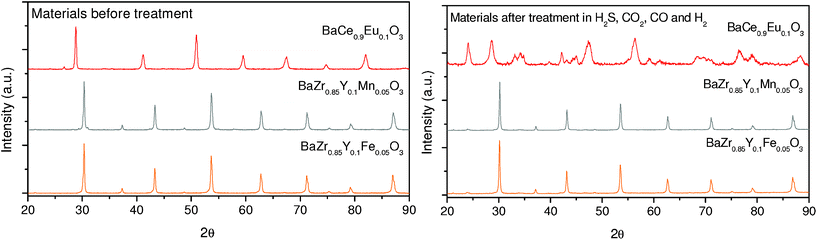 | ||
| Fig. 12 XRD diffraction patterns of BaZr0.85Y0.1Fe0.05O3-δ and BaZr0.8Y0.15Mn0.05O3-δ before and after the stability test in a mixture of 115ppm H2S, 4.43% CO2, 2.12% CO and 92.09% H2, carried out for 40 h at 500 °C and 30 bars. The XRD diffraction pattern of BaCe0.9Eu0.1O3-δ was added for comparison. | ||
In order to ensure the stability of zirconates in CO2-containing atmospheres, the different materials were additionally studied by thermogravimetry (TG). TG curves (Fig. 13) were recorded for BaZr0.9Y0.1O3-δ, BaZr0.8Y0.15Mn0.05O3-δ and BaCe0.9Yb0.1O3-δ under continuous gas flow of a mixture of 5% CO2, while the temperature was raised up to 1000 °C. For all studied samples, a small weight loss was observed with temperature increase up to 500 °C, which may be attributed to sample dehydration and slight oxygen release, as also discussed in the conductivity measurements section. BaCe0.9Yb0.1O3-δ shows a gradual weight gain recorded above 600 °C that can be ascribed to the formation of carbonates.40,41 Further heating induced weight loss at 950 °C implies that the reverse reaction may have taken place, leading to release of CO2. No reaction with CO2 was observed in the TG curve of BaZr0.9Y0.1O3-δ and BaZr0.8Y0.15Mn0.05O3-δ.
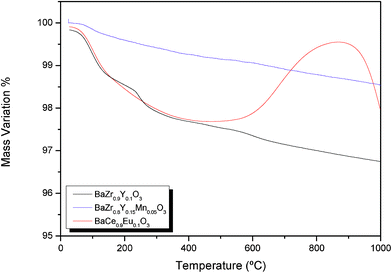 | ||
| Fig. 13 TG measurements of BaZr0.9Y0.1O3-δ and BaZr0.8Y0.15Mn0.05O3-δ in 5% CO2 in Ar. TG data for BaCe0.9Yb0.1O3-δ were plotted for comparison. | ||
Finally, Raman spectroscopy was used as a concluding technique to confirm the stability of the developed compounds in CO2, as it can be used for in situ detection of carbonate formation on the perovksite surface, which are hardly detected by XRD. Results recorded for the BaZr0.8Y0.15Mn0.05O3-δ sample as sintered in air, and after exposure to a continuous flow of 15% CO2 in Ar at 750 °C for 3 h, are presented in Fig. 14. From the experiment carried out, it can be ascertained that in this set of test conditions no formation of carbonate was observed, since the same structural peaks appear as seen in the graph. In contrast, the formation of BaCO3 was observed when Yb-doped BaCeO3 was exposed to the same temperature-programmed carbonation treatment as reported in ref. 34.
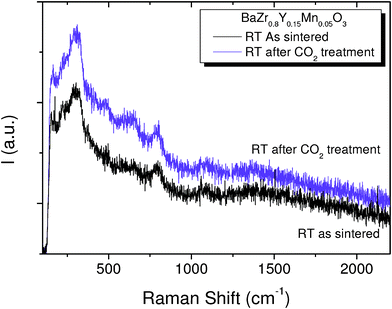 | ||
| Fig. 14 Raman spectroscopy analysis carried out for the BaZr0.8Y0.15Mn0.05O3-δ sample: the black curve is for the as-sintered sample and the blue curve is for the sample being treated in a continuous flow of 15% CO2 in Ar at 750 °C for 3 h. | ||
4. Conclusions
The present study shows structural and transport properties of Pr-, Fe-, and Mn-doped BaZr1-x-yYxMyO3-δ compounds. Once demonstrated that the Mn-dopant leads to significant improvement in the total conductivity of the parent compound, BaZr0.8Y0.15Mn0.05O3-δ was selected for carrying out the hydrogen permeation measurements. This compound exhibited the highest total and protonic conductivities at elevated temperatures under reducing atmospheres while a particular redox behavior related to Mn species is observed under these conditions. The hydrogen permeation flow obtained through the BaZr0.8Y0.15Mn0.05O3-δ membrane was higher than previously reported for the cerates42,43 and also comparable to other compounds, as for example solid solutions of cerates and zirconates.44 In addition, the stability of BaZr0.8Y0.15M0.05O3-δ under operation-relevant atmospheres was studied through several techniques and the results showed unambiguously that this material has a high stability in both CO2 and H2S-containing operation environments. Finally, it can be pointed out that, from the developed materials in the series, BaZr1-x-yYxMyO3-δ, BaZr0.8Y0.15M0.05O3-δ is a promising candidate for industrial hydrogen-oriented applications, and competitive hydrogen flow of around 7 ml min−1 cm−2 at 900 °C could be achieved using 20 μm-thick membranes. Therefore, supported thin layer membranes should be developed to minimize the required membrane area for the targeted industrial applications.Acknowledgements
Financial support by the Spanish Ministry for Economics and Competitiveness (JAE-Pre 08-0058, ENE2008-06302 and ENE2011-24761 grants) and the Helmholtz Association of German Research Centers through the Helmholtz Alliance MEM-BRAIN (Initiative and Networking Fund) is kindly acknowledged. Dr M. Ivanova thanks the Northern European Innovative Energy Research Program N-INNER (grant no. 09-064274) and the German Federal Ministry of Education and Research (BMBF) for supporting the N-INNER Project “Novel High Temperature Proton and Mixed-Proton Electron Conductors for Fuel Cells and H2-separation membranes” (Contract 03SF0330).References
- T. Norby and Y. Larring, Solid State Ionics, 2000, 136, 139 CrossRef.
- H. Iwahara, Solid State Ionics, 1995, 77, 289 CrossRef CAS.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- K. H. Ryu and S. M. Haile, Solid State Ionics, 1999, 125, 355 CrossRef CAS.
- P. Babilo and S. M. Haile, J. Am. Ceram. Soc., 2005, 88, 2362 CrossRef CAS.
- J. M. Serra and W. A. Meulenberg, J. Am. Ceram. Soc., 2007, 90, 2082 CrossRef CAS.
- W. A. Meulenberg, J. M. Serra and T. Schober, Solid State Ionics, 2006, 177, 2851 CrossRef CAS.
- S. Ricote, G. Caboche and O. Heintz, J. Appl. Electrochem., 2009, 39, 553 CrossRef CAS.
- J. Tong, D. Clark, L. Bernau, A. A. Subramaniyan and R. O'Hayre, Solid State Ionics, 2010, 181, 496 CrossRef CAS.
- S. J. Song, T. H. Lee, L. Cheng, S. E. Dorries and U. Balachandran, US Pat. US2010/0031822 A1 to U Chicago Argonne, LLC Search PubMed.
- S.-J. Song, E. D. Wachsman, J. Rhodes, H.-S. Yoon, K.-H. Lee, G. Zhang, S. E. Dorris and U. Balachandran, J. Mater. Sci., 2005, 40, 4061 CrossRef CAS.
- H. Matsumoto, T. Shimura, T. Higuchi, H. Tanaka, K. Katahira, T. Otake, T. Kudo, K. Yashiro, A. Kaimai, T. Kawada and J. Mizusakia, J. Electrochem. Soc., 2005, 152, A488 CrossRef CAS.
- H. Matsumoto, T. Shimura, H. Iwahara and K. Katahira, US Pat. US2006/0157674 A1 to Nagoya University Search PubMed.
- X. Wei, J. Kniep and Y. S. Lin, J. Membr. Sci., 2009, 345, 201 CrossRef CAS.
- J. Kniep and Y. S. Lin, Ind. Eng. Chem. Res., 2010, 49, 2768 CrossRef CAS.
- G. C. Mather, D. Poulidi, A. Thursfield, M. J. Pascual, J. R. Jurado and I. S. Metcalfe, Solid State Ionics, 2010, 181, 230 CrossRef CAS.
- M. Y. Cai, H. X. Luo, Z. Li, A. Feldhoff, J. Caro and H. H. Wang, Chin. Chem. Lett., 2008, 19, 1256 CrossRef CAS.
- T. Oh, H. Yoon, J. Li and E.D. Wachsman, J. Membr. Sci., 2009, 345, 1 CrossRef CAS.
- Z. Sun, E. Fabbri, L. Bi and E. Traversa, Phys. Chem. Chem. Phys., 2011, 13, 7692 RSC.
- M. Shirpour, R. Merkle and J. Maier, Solid State Ionics, 2011 DOI:10.1016/j.ssi.2011.09.006.
- D. Pergolesi, E. Fabbri, A. D'Efifanio, E. Di Bartolomeo, A. Tebano, S. Sanna, S. Licoccia, G. Balestrino and E. Traversa, Nat. Mater., 2010, 9, 846 CrossRef CAS.
- H. Iwahara, T. Esaka, H. Uchida and N. Maeda, Solid State Ionics, 1981, 3, 359 CrossRef.
- T. Norby and Y. Larring, Solid State Ionics, 1995, 77, 147 CrossRef.
- H. G. Bohn and T. Schober, J. Am. Ceram. Soc., 2000, 83, 768 CrossRef CAS.
- S. Ricote, N. Bonanos, H. J. Wang and B. A.Boukamp, Solid State Ionics, 2012, 213, 36 CrossRef CAS.
- S. Ricote, N. Bonanos, A. Manerbino and W. G. Coors, Int. J. Hydrog. Energy, in press DOI:10.1016/j.ihydene.2011.08.118.
- M. Shirpour, B. Rahmati, W. Sigle, P. A. van Aken, R. Merkle and J. Maier, J. Phys. Chem. C, 2012, 116, 2453 CAS.
- I. Levin, T. G. Amos, S. M. Bell, L. Farber, T. A. Vanderah, R. S. Roth and B. H. Toby, J. Solid State Chem., 2003, 175, 170 CrossRef CAS.
- W. Wang and A. V. Virka, J. Power Sources, 2005, 142, 1 CrossRef CAS.
- A. S. Nowik and Y. Du, Solid State Ionics, 1997, 97, 17 CrossRef.
- K. D. Kreuer, St. Adams, W. Munch, A. Fuchs, U. Klock and J. Maier, Solid State Ionics, 2001, 145, 295 CrossRef CAS.
- W. P. Stege, L. E. Cadus and B. P. Barbero, Catal. Today, 2011, 172, 53 CrossRef CAS.
- S. Ponce, M. A. Peña and J. L. G. Fierro, Appl. Catal., B, 2000, 24, 193 CrossRef CAS.
- S. Escolástico, C. Solís and C. J. M. Serra, Int. J. Hydrogen Energy, 2011, 36, 11946 CrossRef.
- H. Matsumoto, T. Shimura, H. Iwahara, T. Higuchi, K. Yashiro and A. Kaimai, J. Alloys Compd., 2006, 408, 456 CrossRef.
- W. A. Meulenberg, M. Ivanova, St. Roitsch and J. M. Serra in Advanced membrane science and technology for sustainable energy and environmental applications, Woodhead Publishing Series in Energy No. 25, ISBN 978-1-84569-969-7, ed. Angelo Basile and Suzana Pereira Nuñes, Woodhead Publishing Limited, Cambridge, UK, 2011, ch. 17, Part IV, pp. 541–567 Search PubMed.
- S. Zhan, X. Zhu, B. Ji, W. Wang, X. Zhang, J. Wang, W. Yang and L. Lin, J. Membr. Sci., 2009, 340, 241 CrossRef CAS.
- S. Hamakawa, L. Li, A. Li and E. Iglesia, Solid State Ionics, 2002, 148, 71 CrossRef CAS.
- S. Baumann, J. M. Serra, P. Lobera, S. Escolástico, F. Schulze-Küppers and W. A. Meulenberg, J. Membr. Sci., 2011, 377, 198 CrossRef CAS.
- K. H. Ryu and S.M. Haile, Solid State Ionics, 1999, 125, 355 CrossRef CAS.
- M. J. Scholten, J. Schoonman, J. C. Vanmiltenburg and H. A. J. Oonk, Solid State Ionics, 1993, 61, 83 CrossRef CAS.
- X. Wei, J. Kniep and Y. S. Lin, J. Membr. Sci., 2009, 345, 201 CrossRef CAS.
- S. J. Song, E. D. Wachsman, J. Rhodes, S. E. Dorris and U. Balachandran, Solid State Ionics, 2004, 167, 99 CrossRef CAS.
- J. Kniep and Y. S. Lin, Ind. Eng. Chem. Res., 2010, 49, 2768 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20214j/ |
‡  |
| This journal is © The Royal Society of Chemistry 2012 |