The fluorine effect: photophysical properties of borondipyrromethene (bodipy) dyes appended at the meso position with fluorinated aryl groups†
Mohammed A. H.
Alamiry
a,
Andrew C.
Benniston
*a,
Jerry
Hagon
a,
Thomas P. L.
Winstanley
a,
Helge
Lemmetyinen
b and
Nikolai V.
Tkachenko
b
aMolecular Photonics Laboratory, School of Chemistry, Newcastle University, Newcastle upon Tyne, UK. E-mail: andrew.benniston@ncl.ac.uk.; Fax: +44 191 222 6929; Tel: +44 191 222 5706
bDepartment of Chemistry and Bioengineering, Tampere University of Technology, PO Box 541, 33101, Finland
First published on 27th March 2012
Abstract
A small series of partially alkylated Bodipy dyes were prepared containing Fn-aryl (n = 1,2,3,5) groups at the meso position. The effect of increasing the number of fluorines, and their position in the aryl ring, on the electrochemical and photophysical properties of the dyes is discussed. The highly electron withdrawing pentafluoroaryl group makes reduction of the Bodipy segment especially easy when compared to the basic phenylene derivative. High quantum yields of fluorescence in toluene solution are seen for derivatives with fluorine(s) substituted in the ortho position of the meso aryl group. This effect is also mirrored in the fluorescence lifetimes for the molecules. Pressure dependent fluorescence measurements carried out reveal subtle differences in the behaviour for the series of Bodipy derivatives.
Introduction
The benefits to introducing a single fluorine atom, or multiple fluorines, into organic molecules are well documented.1 The prime example is for the field of medicinal biosteres, since replacement of hydrogen for fluorine retards metabolic oxidation.2 Because of the small difference in size between fluorine and hydrogen the molecular topology is not altered greatly, and hence the efficacy of biological action is not affected.3 Evidently, the highly electron withdrawing nature of fluorine affects the electron affinic nature of the group to which it is attached. Moreover, in aromatic rings the number and substitution pattern influences the energy of the σ and π frameworks.4 In poly-conjugated systems, for example, the introduction of fluorine atoms is known to lower both the HOMO and LUMO levels, and to facilitate electron injection.5 In comparison to the p-type behaviour for non-fluorinated materials, the electron deficient nature of fluorine containing compounds makes them ideal n-type semiconductors.6 The insertion of fluorine into polythienothiophene-co-benzodithiophenenes in photovoltaic cells was shown to increase power conversion efficiency.7 There is clear precedent that fluorinated compounds have a benefit over their hydrogen-based counterparts in diverse applications.8Considering the electronic perturbation by fluorination, it is not too surprising that several studies were conducted into studying what can be termed the fluorine effect.9 As a follow on to these studies we discuss the photophysical properties of several meso aryl borondipyrromethene (Bodipy) compounds, and the effect of fluorine substitution into the aryl ring. The main impetus for this work came from the reported observation of near unity quantum yield of fluorescence in solution for the pentafluoroaryl derivative, BD-F5. The high fluorescence yield represents a significant enhancement with respect to the basic phenylene compound, BD-Ph (Fig. 1).10 Our intention was to try and understand this result through a systematic study, by reducing the number of fluorines in the aryl ring, and the substitution pattern for mono-fluorinated derivatives. It became clear that the important factor was not the number of fluorines but, in fact, substitution at the ortho position in the aryl ring.
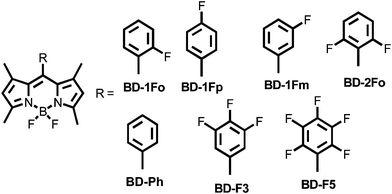 | ||
| Fig. 1 Molecular formulae of Bodipy derivatives discussed in the text. | ||
Synthesis and characterisation
The target compounds shown in Fig. 1 were prepared by standard procedures,11 and purified rigorously to remove spurious fluorescent impurities. The compounds were characterised by standard analytical techniques including 1H, 13C, 19F, 11B NMR spectroscopies, mass spectrometry and melting points. X-Ray structure determinations were carried out on all compounds and will be reported elsewhere. All data were fully consistent with the proposed structures. The one especially interesting case pertained to compound BD-1Fo, and the 19F NMR spectrum which displayed a multiplet (Fig. 2) rather than the standard quartet. The pattern was analysed as two doublet of quartets, owing to the inequivalence of the two fluorines in the BF2 unit. Rotation of the meso monofluorinated aryl group must be restricted by the flanking 1,7-methyl units to cause the two fluorines to reside in different chemical environments. Even at elevated temperatures in d10-o-xylene there was no sign of coalescence for the fluorine signals. The rate for rotation of the aryl group in BD-1Fo must be < 1 s−1 at room temperature. Such a slow rate is presumably the same for the other ortho fluorine and hydrogen derivatives considering the similar size of the fluorine and hydrogen atoms. Restricted rotation behaviour was reported recently for a phenanthrene-based Bodipy derivative.12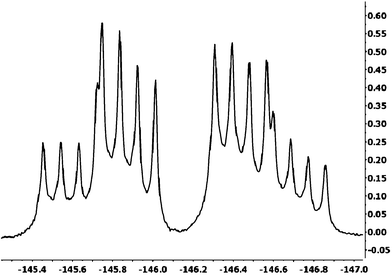 | ||
| Fig. 2 19F NMR spectrum for BD-1Fo recorded in CDCl3 at room temperature. | ||
Molecular modelling
To better comprehend the effect of fluorine substitution for the Bodipy derivatives we undertook a comprehensive computational study using Gaussian 03.13 To reduce run time in the first instance, the ground-state energy-minimised structures were calculated in the gas phase at a low-level of theory (Hartree–Fock). Further refinement and optimisation on structures were then undertaken using DFT and B3LYP and the 6-311G++ basis set. No higher level of calculation was deemed necessary, considering the purpose was to compare properties of the molecules. The energy-minimised structures for the Bodipy compounds are collected in the ESI.† It is noted that for each structure the aryl group is orthogonal to the dipyrrin plane, owing to unfavourable steric interactions between the ring and the two flanking methyl groups. The calculated electrostatic potentials for the derivatives are shown in Fig. 3. Not surprisingly, the highly electron withdrawing fluorines attached to the boron atom polarise the electrostatic potential at this site. When compared to basic BD-Ph the introduction of fluorine(s) into the aryl ring, as expected, distorts the electron distribution in the aryl ring. There is no evidence that the pentafluoroaryl group is especially electron withdrawing, and that it pulls electron density out of the dipyrromethene unit.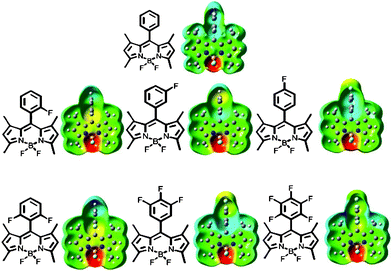 | ||
| Fig. 3 Illustrations of the total electrostatic potential mapped onto the isosurface of the molecular orbital for the Bodipy derivatives. Note: the more negative the value of the potential the redder the colour. | ||
The subtle differences between the Bodipy compounds is best appreciated by comparison of the HOMO and LUMO energies. For all the cases, the overall appearance of the orbitals is similar, with the HOMO residing primarily on the two pyrrole-like groups with little electron density located at the meso position. The LUMO is more spread out over the entire dipyrrin backbone (see ESI†). The results are in good agreement with previously published calculations on similar Bodipy derivatives.14 Collected in Table 1 are the values for energies of the HOMO and LUMO levels and their separation in energy (ΔE). Compared to the basic phenylene derivative the introduction of fluorine into the aryl ring lowers both the HOMO/LUMO levels on the Bodipy group. The effect of mono-fluorination on the molecular orbital energies is similar for the meta and para derivatives. It is also noticeable that the HOMO/LUMO levels are shifted relative to each other such that the ΔE values are the same as BD-Ph. In comparison, ΔE values for both BD-1Fo and BD-2F are reduced relative to the standard. Even though the HOMO/LUMO levels for BD-3F are lowered in energy (cf.BD-2F), ΔE is more comparable to BD-Ph. It is apparent that any electronic effect is greater for fluorine(s) in the ortho position of the aryl ring. At the extreme case for BD-5F some cumulative electronic effect emerges since ΔE is the smallest out of all the compounds. Since ΔE is related to the colour of the compounds a simple prediction for the absorption maxima ordering is BD-1Fm ∼ BD-1Fp ∼ BD-Ph < BD-3F < BD-1Fo < BD-2F< BD-5F. As discussed later this prediction is not too dissimilar to the actual result.
| Compound | E HOMO b | E LUMO b | ΔE/eVc |
|---|---|---|---|
| a Calculated for gas-phase energy-minimised structures. b Energy in Hartrees. c Energy gap for HOMO–LUMO. | |||
| BD-Ph | −0.213 | −0.103 | 2.99 |
| BD-1Fo | −0.214 | −0.106 | 2.94 |
| BD-1Fm | −0.218 | −0.108 | 2.99 |
| BD-1Fp | −0.218 | −0.108 | 2.99 |
| BD-2F | −0.215 | −0.108 | 2.91 |
| BD-3F | −0.227 | −0.118 | 2.97 |
| BD-5F | −0.228 | −0.123 | 2.86 |
Electrochemistry
The redox properties of the fluorinated derivatives were studied under identical conditions by cyclic voltammetry in dry CH3CN containing 0.2 M TBATFB. For comparison purposes the redox behaviour of BD-F5 and BD-Ph are discussed in detail. The cyclic voltammograms for the two derivatives are shown in Fig. 4. At positive potentials both compounds display quasi-reversible waves at E1/2 = +1.33 V and E1/2 = + 1.22 V for BD-Ph and BD-F5, respectively. Both waves are associated with oxidation of the Bodipy group. The electron withdrawing pentafluoroaryl group clearly makes electron removal from the HOMO more difficult in BD-F5 when compared to the standard. On reductive scanning the ‘fluorine effect’ is again very obvious. Reversible waves are located at E1/2 = −1.08 V and E1/2 = −0.83 V for BD-Ph and BD-F5, respectively, and are associated with reduction of the Bodipy group. The large anodic shift for BD-F5 is again supportive of the capacity for the electron withdrawing pentafluoroaryl group to stabilise the radical anion. The electrochemical behaviour of the other derivatives is understood in terms of the above model. Collected in Table 2 are the electrode potentials for the other compounds.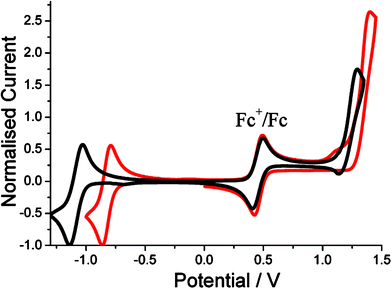 | ||
| Fig. 4 Cyclic voltammograms for BD-Ph (black) and BD-5F (red) recorded in CH3CN vs. Ag/AgCl. | ||
Absorption and fluorescence
The ambient temperature electronic absorption spectra for the derivatives were recorded in dilute toluene. Profiles for BD-F5 and BD-1Fo are shown in Fig. 5. The typical sharp So–S1 electronic transition for BD-F5, situated at λABS = 509 nm, is red-shifted by some 11 nm with respect to BD-1Fo. The molar absorption coefficient (εMAX) of 76,000 M−1 cm−1 for BD-F5 is reduced slightly when compared BD-1Fo. In comparison, the much broader band associated with the S0–S2 electronic transition at around 350 nm is hardly affected by the increase in the number of fluorine atoms in the aryl ring. The observed red shift is accounted for by the decrease in the HOMO–LUMO gap. Such an effect is consistent with both the DFT computed and electrochemistry calculated ΔE values. Both compounds are extremely fluorescent in fluid toluene solution at room temperature. Emission profiles, with peak maxima (λFLU), for both compounds are fairly good mirror images of their long-wavelength absorption bands. The magnitudes of the Stokes' shifts (SS) are consistent with those found for Bodipy derivatives.15 The quantum yields of fluorescence (φFLU) are 1.0 and 0.98 for BD-F5 and BD-1Fo, respectively. Fluorescence lifetimes (τs) as measured by single-photon-counting were strictly mono-exponential, but the lifetime for BD-5F is appreciably longer compared to BD-1Fo. There is a small difference in the radiative rate constants (kRAD = φFLU/τs) for the two derivatives. Photophysical data collected for the other derivatives are collected in Table 3. By comparison of the data it follows that: (i) The increase in λABS follows the order BD-Ph < BD-1Fm ∼ BD-1Fp < BD-3F ∼ BD-1Fo < BD-2F < BD-5F. (ii) The largest SS is seen for BD-5F but there is no obvious overall trend in the series. (iii) The smallest kNR values are observed for the Fn-aryl Bodipy derivatives with fluorine(s) in the ortho position. (iv) The meta or para substitution pattern for fluorines does not affect significantly the fluorescence lifetimes.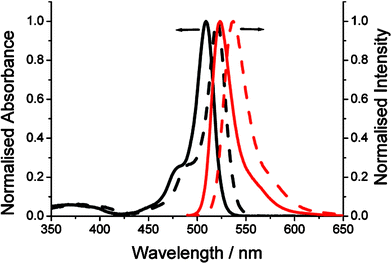 | ||
| Fig. 5 Ambient temperature absorption (black solid = BD-1Fo, black dash = BD-5F) and fluorescence (red solid = BD-1Fo, red dash = BD-5F) spectra recorded in dilute toluene. | ||
| Comp. | λ ABS b nm | ε MAX M−1 cm−1 | λ FLU b nm | SSc cm−1 | φ FLU d | τ s e ns | k RAD 108 s−1 | k NR 106 s−1 |
|---|---|---|---|---|---|---|---|---|
| a Measured in CH3CN and taken from ref. 22. b ± 2 nm. c ± 25 cm−1. d ± 10%. e ± 5%. | ||||||||
| BD-Ph a | 503 | 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
515 | 464 | 0.71 | 3.9 | 1.82 | 76.4 |
| BD-1Fo | 509 | 81000 |
523 | 526 | 0.98 | 4.9 | 2.0 | 4.1 |
| BD-1Fm | 505 | 98000 |
520 | 571 | 0.88 | 3.5 | 2.5 | 35.7 |
| BD-1Fp | 505 | 98000 |
520 | 571 | 0.88 | 3.5 | 2.5 | 35.7 |
| BD-2F | 515 | 77000 |
530 | 549 | 0.99 | 5.1 | 1.94 | 2.0 |
| BD-3F | 509 | 98000 |
523 | 526 | 0.91 | 3.6 | 2.5 | 27.8 |
| BD-5F | 520 | 76000 |
537 | 609 | 1.0 | 5.7 | 1.75 | <0.40 |
Plainly, what is the origin for reduction in non-radiative rates for the ortho Fn-aryl Bodipy derivatives? Work has previously shown that modest distortion of the dipyrrin backbone is linked to deactivation of the Bodipy S1 state.16 The greater the bowing of the molecular framework the more efficient is non-radiative decay. The effect is especially noticeable in Bodipy derivatives without the methyl substituents (i.e., the so called molecular rotors).17 For such systems rotation of the meso aryl group is relatively unhindered and the quantum yield of fluorescence is low. Free rotation of the aryl group in the compounds discussed here is very unlikely, even for excited state structures. The rigidity of the carbon framework in alkanes is enhanced through fluorination.18 There is no obvious reason why the dipyrrin framework is strengthened by a similar effect in the cases discussed here. One point to note is the previously reported differences in φFLU for the methyl-aryl analogues of BD-1Fo and BD-1Fp (i.e., fluorine is replaced by methyl).19,20 The φFLU is noticeably greater for the methyl ortho derivative. It would appear that a steric rather than an electronic effect is controlling non-radiative decay. The increases in the van der Waals volume with exchange of phenyl for the mono- (BD-1Fo) and di-fluorinated (BD-2F) aryl units are 7% and 14%, respectively. Instead of full rotation of the aryl unit it presumably rocks back and forth rapidly between two extreme points defined by an angle θ. For the ortho-fluorinated derivatives θ must be reduced with respect to the case for BD-Ph. One suggestion is that the extent of distortion of the dipyrrin group is linked to the angle θ. That is, for small values of θ the distortion of the dipyrrin is minor, and is hence associated with high fluorescence quantum yields. One contribution which cannot be discounted is, of course, from the so termed “ortho effect”. This steric term has been attributed to many tentative components which are not easy to diagnose: primary steric effect, steric inhibition to resonance, short range field effect, hydrogen bonds and steric inhibition to solvation.21
Pressure-dependent fluorescence studies
If non-radiative decay was linked to distortion of the Bodipy S1 structure, one expectation was that fluorescence yield might depend on applied pressure. Fluorescence spectra were recorded for all the compounds in methylcyclohexane (MChx) up to an applied pressure of 550 MPa. The responses for BD-1Fo and BF-5F are depicted in Fig. 6. It is noted that MChx undergoes a slight phase change at around 280 MPa, owing to a switch from axial to equatorial conformations so as to reduce the molar volume.23 For BD-1Fo, application of pressure causes a small increase in the apparent fluorescence yield (∼6%)24 up to around the point where the molar volume of the solvent changes. After this the fluorescence yield decreases steadily. There is a concomitant red-shift of 5 nm in λem. In comparison, for BD-5F the increasing pressure is again accompanied by a red shift (5 nm), but the fluorescence yield decreases progressively from the onset. The behaviour for BD-2F is very similar to the case for BD-5F. In contrast, increasing the pressure for BD-1Fm, BD-1Fp and BD-3F leads to a significant increase in fluorescence yield over the entire pressure range (see ESI†). Releasing the pressure restores the original spectra in all the cases.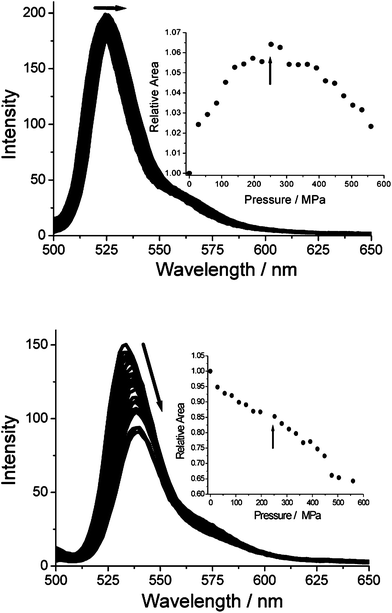 | ||
| Fig. 6 Effect of applied pressure on the uncorrected fluorescence spectra recorded for BD-1Fo (top) and BD-5F (bottom) in dilute MChx. Insert shows the change in relative peak area with respect to the applied pressure. The arrows show the pressure at which the solvent undergoes a phase change. | ||
Up to a pressure of 550 MPa the decrease in volume of MChx is around 17%.25 Over the same pressure range the increase in the refractive index (n) is approximately 12%.25 Any small increase in fluorescence yield can be accounted for by the increase in solute concentration. The small red shift in λem is due to the small change in polarizability (eqn (1)) of the solvent, as witnessed for previous studies.26
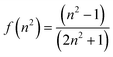 | (1) |
It appears that the non-radiative decay pathway for the ortho Fn-aryl Bodipy compounds is enhanced under high pressure. As the applied pressure increases the distortion of the dipyrrin unit must increase dramatically, especially for BD-2F and BD-5F. The aryl group probably tries to adopt a more planar arrangement with respect to the dipyrrin unit. Presumably, two ortho fluorine atoms clash more severely with the two methyl groups than do hydrogen atoms.
Conclusions
Previously, the Hammett constant (σ) for the pentafluoroaryl group has been estimated to be 1.19 from computational calculations.27 Such a high σ value is consistent with the group acting as a stronger electron withdrawing group than, for example, SO2C(CF3)3.28 Our electrochemical finding is certainly consistent with the highly electron withdrawing nature of the group. Perturbation of the LUMO on the Bodipy by the pentafluoroaryl unit is perhaps not that surprising: the orbital contribution for the LUMO at the meso site is significant. It might appear that to increase the fluorescence lifetime for a Bodipy derivative, meso attachment of a highly electron withdrawing group is important. Very subtle ortho steric effects also play just as an important role, however. To produce an easy to reduce Bodipy which is very fluorescent, a fully substituted highly electron deficient aryl group appears to be the best option. The pentakiscyano- or pentakistrifluoromethyl-benzene units appear to be interesting candidates, and we expect to prepare and study such compounds in the near future.Experimental
All chemicals were purchased from commercial sources and used as received unless otherwise stated. Basic solvents for synthesis were dried using literature methods. Solvents for spectroscopic investigations were of the highest purity available. DCM = dichloromethane, DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, TFA = trifluoroacetic acid, TBATFB = N-tetrabutylammonium tetrafluoroborate.Preparation of BD-F5
A 500 mL round bottomed flask was charged with DCM (250 mL), and purged with N2 for 1 h. To the flask were added 2,3,4,5,6- pentafluorobenzaldehyde (0.63 mL, 5.1 mmol), 2,4-dimethyl pyrrole (1.10 mL, 10.7 mmol), TFA (3 drops), and the reaction mixture was stirred at room temperature until all the aldehyde was consumed. Upon completion of the reaction DDQ (1.16 g, 5.1 mmol) was added, and the reaction mixture was stirred at room temperature for 12 h. After this time, N,N-diisopropylethylamine (5.03 mL, 29.1 mmol) and BF3·Et2O (5.03 mL, 40.8 mmol) were added and the reaction mixture was monitored by tlc until loss of the initially produced material. The organic solvent mixture was extracted with water (3 × 100 mL) and brine (200 mL). The aqueous layer was backwashed with DCM (100 mL), and the organic portions were combined and dried over MgSO4. After filtration the solvent was removed under reduced pressure. The crude product was purified via column chromatography (silica gel, DCM: petrol gradient elution). Yield: 190 mg, 0.45 mmol, 9%. 1H NMR (CDCl3, 400 MHz): δ = 6.05 (s, 2H), 2.56 (s, 6H), 1.60 (s, 6H). 13C NMR (CDCl3, 100 MHz): δ = 157.86, 145.09, 143.09, 143.08 141.59, 139.34, 137.25, 131.07, 122.82, 122.33, 109.60, 14.85, 13.65. 11B NMR (CDCl3, 128 MHz): δ = −0.29 (t, J = 33.2 Hz). 19F NMR (CDCl3, 376 MHz): δ = −139.14 (2F), −146.10 (2F), -150.37 (1F), −159.18, −159.56 (2F). IR 2923 cm−1 (C–H) 1540 cm−1, 1496 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN), 1160 cm−1 (B–F). MP = 237 °C.
C, CN), 1160 cm−1 (B–F). MP = 237 °C.
The other fluorinated derivatives were prepared in a similar manner to that above.
BD-1Fo
15% yield. 1H NMR (CDCl3, 400 MHz): δ = 7.46–7.37 (m, 1H), 7.21 (dd, J = 6.1, 1.7 Hz, 2H), 7.13 (t, J = 8.9 Hz, 1H), 5.93 (s, 2H), 2.49 (s, 6H), 1.39 (s, 6H). 13C NMR (CDCl3, 100 MHz): 189.76, 160.42, 157.96, 142.74, 134.63, 131.43, 130.47, 125.12, 122.79, 122.62, 116.53, 14.70, 13.86. 11B NMR (CDCl3, 128 MHz): δ = −0.19. 19F NMR (CDCl3, 376 MHz): δ = −114.36 (1F), −146.16 (2F). MP = 175–176 °C. TOF-MS calc. for C19H18BF3N2 = 342, fnd. 365 (M + Na+), 707 (2 M + Na+). IR 2979 cm−1 (C–H) 1545 cm−1, 1509 cm−1 (CC, CN), 1187 cm−1 (B–F).
BD-1Fp
10% yield. 1H NMR (CDCl3, 400 MHz): δ = 7.25 (dd, J = 8.8, 5.3 Hz, 2H), 7.19 (t, J = 8.6 Hz, 2H), 5.97 (s, 2H), 2.53 (s, 6H), 1.38 (s, 6H). 13C NMR (CDCl3, 100 MHz): δ = 163.25, 155.87, 143.54, 131.00, 130.04, 121.48, 166.47, 14.67, 14.60. 11B NMR (CDCl3, 128 MHz): δ = −0.20. 19F NMR (CDCl3, 376 MHz): δ = −111.72, −146.07. MP = 170–172 °C. TOF-MS calc. for C19H18BF3N2 = 342, fnd. 365 (M + Na+), 707 (2 M + Na+). IR 2926 cm−1 (C–H), 1547 cm−1, 1508 cm−1 (CC, CN), 1186 cm−1 (B–F).
BD-1Fm
11% yield. 1H NMR (CDCl3, 400 MHz): δ = 7.46 (td, J = 8.0, 5.8 Hz, 1H), 7.17 (tdd, J = 8.6, 2.8, 1.1 Hz, 1H), 7.08 (dt, J = 7.6, 1.3 Hz, 1H), 7.02 (ddd, J = 8.9, 2.6, 1.5 Hz, 1H), 5.93 (s, 2H), 2.49 (s, 6H), 1.35 (s, 6H). 13C NMR (CDCl3, 100 MHz): δ = 164.35, 162.37, 156.05, 143.01, 139.71, 137.14, 131.04, 124.11, 121.55, 116.02, 115.57, 14.69, 14.44. 11B NMR (CDCl3, 128 MHz): δ = −0.22. 19F NMR (CDCl3, 376 MHz): δ = −111.38, −146.17. MP = 156–158 °C. TOF-MS calc. for C19H18BF3N2 = 342, fnd. 365 (M + Na+), 707 (2 M + Na+). IR 2929 cm−1 (C–H), 1540 cm−1, 1505 cm−1 (CC, CN), 1185 cm−1 (B–F).
BD-2F
10% yield. 1H NMR (CDCl3, 400 MHz): δ = 7.56–7.35 (m, 1H), 7.13–6.94 (m, 2H), 6.00 (s, 2H), 2.55 (s, 6H), 1.54 (s, 6H). 13C NMR (CDCl3, 100 MHz): δ = 161.05, 158.57, 156.55, 142.19, 131.67, 131.57, 131.33, 127.37, 121.66, 112.31, 112.06, 14.76, 13.40. 11B NMR (CDCl3, 376 MHz): δ = −0.22. 19F NMR (CDCl3, 376 MHz): δ = −110.20–112.68 (m), −146.0. MP = >240 °C. TOF-MS calc. for C19H17BF4N2 = 360, fnd. 360 (M+). IR 2929 cm−1 (C–H), 1547 cm−1, 1508 cm−1(CC, CN), 1186 cm−1 (B–F).
BD-3F
25% yield. 1H NMR (CDCl3, 400 MHz): δ = 6.92 (t, J = 6.9 Hz, 2H), 5.95 (s, 2H), 2.48 (s, 6H), 1.43 (s, 6H). 13C NMR (CDCl3, 100 MHz): δ = 156.85, 150.73, 142.57, 141.73, 141.58, 139.20, 138.89, 137.14, 130.93, 121.93, 113.28, 14.73, 14.67. 11B NMR (CDCl3, 128 MHz): δ = −0.30. 19F NMR (CDCl3, 376 MHz): δ = −131.85, −146.12, −158.37. MP = 181–182 °C. EI-MS calc. for C19H16BF5N2 = 378, fnd. 378 (M+). IR 2929 cm−1 (C–H), 1550 cm−1, 1511 cm−1 (CC, CN), 1189 cm−1 (B–F).
Cyclic voltammetry experiments were performed using a fully automated HCH Instruments Electrochemical Analyzer and a three-electrode set-up consisting of a glassy carbon working electrode, a platinum wire counter electrode and an Ag/AgCl reference electrode. Ferrocene was used an internal standard. All studies were performed in deoxygenated CH3CN containing TBATFB (0.2 M) as background electrolyte. Redox potentials were reproducible to within ±15 mV. All luminescence measurements were made using optically dilute solutions and were corrected for spectral imperfections of the instrument by reference to a standard lamp. Luminescence quantum yields were measured relative to a standard Bodipy dye (φFLU = 0.70)29 using optically matched solutions.
The fluorescence lifetime measurements were carried out using time-correlated single-photon-counting instrument (PicoHarp-300, PicoQuant GmbH). The samples were excited at 483 nm by a pulsed diode laser (LDH-P-C-485, PicoQuant GmbH). The emission was detected by a microchannel plate photomultiplier tube (R3809U-50, Hamamatsu) coupled with a monochromator. The time resolution of the system was 80 ps (FWHM).
High pressure studies were carried out using a custom built rig supplied by Stansted Fluid Power Ltd and used in conjunction with a high-pressure sample holder equipped with optically transparent windows. Fluorescence measurements were made using a low-power laser diode (λmax = 406 nm) to excite the sample, and collecting emission with a focussing lens connected to a fibre optic cable that was fed to a red-sensitive photomultiplier tube by way of a high-radiance monochromator and amplifier. Scattered excitation light was minimized with optical filters. In a typical run, the sample, dissolved in MChx, was pressurized up to ca. 550 MPa. Spectra were measured at regular intervals as the pressure exerted on the sample was released in a controlled manner. Fluorescence background spectra were recorded using solvent only and the final emission spectrum corrected as needed. Several runs were recorded for each sample. In other runs, the spectra were recorded as the pressure was increased progressively. In all cases, the temperature was maintained at 22 °C.
The computational calculations were performed using a 32-bit version of Gaussian03 on a quadruple-core Intel Xeon system with 4 GB RAM. The calculations were run in parallel, fully utilising the multi-core processor. To reduce computational time low-level calculations were carried out to minimise structures using Hartree–Fock and a low-level basis set. Energy-minimised structures were then used to feed high-level DFT calculations starting firstly with B3LYP and the 3-21G basis set. The complexity of the basis set was increased and results from calculations compared. The 6-311G++ basis set was deemed sufficient for comparison purposes of the HOMO and LUMO energies between molecules
References
- J. C. Biffinger, H. W. Kim and S. G. DiMagno, ChemBioChem, 2004, 5, 622 CrossRef CAS.
- H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef.
- C. R. Brundle, M. B. Robin and N. A. Kuebler, J. Am. Chem. Soc., 1972, 94, 1466 CrossRef.
- F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003 RSC.
- (a) J. F. Tannaci, M. Noji, J. McBee and T. D. Tilley, J. Org. Chem., 2007, 72, 5567 CrossRef CAS; (b) S. Chai, S.-H. Wen and K.-L. Han, Org. Electron., 2011, 12, 1806 Search PubMed.
- H. J. Son, W. Wang, T. Xu, Y. Liang, Y. Wu, G. Li and L. Yu, J. Am. Chem. Soc., 2011, 133, 1885 CrossRef CAS.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- (a) F. C. Krebs and H. Spanggaard, J. Org. Chem., 2002, 67, 7185 CrossRef; (b) T. V. Trashakhova, E. V. Nosova, M. S. Valova, P. A. Slepukhin, G. N. Lipunova and V. N. Charushin, Russ. J. Org. Chem., 2011, 47, 753 Search PubMed; (c) T. Mondal and S. Mahapatra, J. Chem. Phys., 2010, 133, 084304 CrossRef CAS; (d) R. Bonnett, A. Harriman and A. N. Kozyrev, J. Chem. Soc., Faraday Trans., 1992, 88, 763 RSC; (e) Y. Yamaguchi, J. Chem. Phys., 2005, 122, 184702 Search PubMed.
- O. Galangau, C. Dumas-Verdes, R. Méallet-Renault and G. Clavier, Org. Biomol. Chem., 2010, 8, 4546 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS.
- A. C. Benniston, A. Harriman, V. Whittle and M. Zelzer, Eur. J. Org. Chem., 2010, 523 CrossRef CAS.
- Gaussian 03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
- J. B. Prieto, F. L. Abeloa, V. M. Martínez and I. L. Arbeloa, Chem. Phys., 2004, 296, 13 CrossRef CAS.
- A. C. Benniston and G. Copley, Phys. Chem. Chem. Phys., 2009, 11, 4124 RSC.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. L. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433 CrossRef CAS.
- M. A. H. Alamiry, A. C. Benniston, G. Copley, K. J. Elliott, A. Harriman, B. Stewart and Y.-G. Zhi, Chem. Mater., 2008, 20, 4024 CrossRef CAS.
- J. S. Jang, M. Blanco, W. A. Goddard III, G. Caldwell and R. B. Ross, Macromolecules, 2003, 36, 5331 CrossRef CAS.
- A. Cui, X. Peng, J. Fan, X. Chen, Y. Wu and B. Guo, J. Photochem. Photobiol., A, 2007, 186, 85 CrossRef CAS.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597 CrossRef CAS.
- S. Böhm, P. Fiedler and O. Exner, New J. Chem., 2004, 28, 67 RSC.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056 CrossRef CAS.
- D. J. Gardiner, G. S. Bassi, G. D. Galvin, A. C. Gorvin and B. P. Straughan, Appl. Spectrosc., 1984, 38, 313 Search PubMed.
- The φFLU for BD-1Fo in MChx is reduced to 0.88 when compared to toluene as solvent. Part of the apparent increase in the fluorescence yield is because of the increase in the optical density at the excitation wavelength.
- D. W. Brazier and G. R. Freeman, Can. J. Chem., 1969, 47, 893 CAS.
- (a) T. Rohand, J. Lycoops, S. Smout, E. Braeken, M. Sliwa, M. Van der Auweraer, W. Dehaen, W. M. De Borggraeve and N. Boens, Photochem. Photobiol. Sci., 2007, 6, 1061 RSC; (b) W. Qin, T. Rohand, M. Baruah, A. Stefan, M. Van der Auweraer, W. Dehaen and N. Boens, Chem. Phys. Lett., 2006, 420, 562 CrossRef CAS; (c) W. Qin, M. Baruah, M. Van der Auweraer, F. C. De Schryver and N. Boens, J. Phys. Chem. A, 2005, 109, 7371 CrossRef CAS; (d) W. Qin, M. Baruah, A. Stefan, M. Van der Auweraer and N. Boens, ChemPhysChem, 2005, 6, 2343 CrossRef CAS; (e) J. Catalán, J. Phys. Chem. B, 2009, 113, 5951 CrossRef CAS; (f) A. Filarowski, M. Kluba, K. Cieślik-Boczula, A. koll, A. Kochel, L. Pandey, W. M. De Borggraeve, M. Van der Auqweraer, J. Catalán and N. Boens, Photochem. Photobiol. Sci., 2010, 9, 996 RSC.
- M. Verma, A. F. Chaudhry and C. J. Fahrni, Org. Biomol. Chem., 2009, 7, 1536 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: Molecular orbital pictures for compounds, fluorescence spectra under applied pressure. See DOI: 10.1039/c2ra20219k |
| This journal is © The Royal Society of Chemistry 2012 |