Designing the head group of CO2-triggered switchable surfactants†
Lauren M.
Scott
,
Tobias
Robert
,
Jitendra R.
Harjani
and
Philip G.
Jessop
*
Department of Chemistry, Queen's University, Kingston, Ontario, Canada K7L 3N6. E-mail: jessop@chem.queensu.ca; Fax: +1 (613) 533-6669; Tel: +1 (613) 533-3212
First published on 27th April 2012
Abstract
The synthesis of a small library of nitrogen-based CO2-switchable cationic surfactants with different head groups is described. These switchable surfactants can be switched on by addition of CO2 and switched off by the removal of CO2. The structure of the switchable head group affects the surfactant's basicity, solubility, heat of protonation, rate and extent of switching, and ability to reversibly stabilize and demulsify oil–water emulsions.
1 Introduction
Industry is highly dependent on amphiphilic structures that can be used as potent surfactants. These compounds allow for the formation of stable emulsions that facilitate many processes such as cleaning, manufacturing and oil recovery. However, in most such processes, the breaking of the emulsion is a necessary later step. Unfortunately, any surfactant added to stabilize an emulsion makes the subsequent demulsification more difficult. Hence, there is a need for surfactants that can be “switched off”. The same arguments apply for surfactant-stabilized suspensions and foams: in most applications it is desirable to switch off the surfactant later in the process.The literature has many examples of switchable surfactants, including some that are switched by oxidation/reduction chemistry or by photochemistry.1 There are, in addition, a very small number of surfactants that can be switched on and off by the addition and removal of CO2. The first report on the effect of CO2 on anionic surfactants (soap) came in 19362 and a practical application of this phenomenon has been described by Moore.3 More recently, we have reported long-tail amidines as CO2-switchable cationic surfactants.4
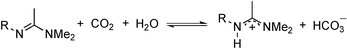 | (1) |
The long-tail acetamidines shown in eqn (1) are demulsifiers of crude-oil–water emulsions when CO2 is absent, but in the presence of CO2 they are converted into long-tail amidinium bicarbonate salts, which serve as emulsion-stabilizing, switchable surfactants. It has been previously established that this conversion results in emulsion stabilization, that it can be reversed, and that the compounds can be repeatedly switched between the two forms.5 We have also reported the effect of structural variations in the tail group on the surfactant's ecotoxicity6 and its ability to stabilize heavy crude-in-water emulsions.7 The acetamidine head group was chosen in our original study because of its significant basicity. A CO2 pressure of 1 atm is sufficient to drive the conversion of the acetamidine to its bicarbonate form almost to completion, despite the relative weakness of formed carbonic acid. The strong demulsifying ability of the acetamidines in their neutral form was an unexpected advantage. We do not expect that alkylacetamidines are the only compounds that can serve as CO2-triggered switchable cationic surfactants; Qin et al. have reported that a long-tail guanidine can function similarly.8 In this paper, we describe the effect of structural variations of the head group on the chemistry of CO2-switchable cationic surfactants.
2 Experimental section
2.1 Materials
Carbon dioxide (Praxair, 99.998%), nitrogen (Praxair, 99.998%), and argon (Praxair, 99.998%) were used as received. North Sea crude oil (API 19.5°, 129 cSt viscosity at 40 °C, 1.65 mgKOH/g total acid number) was obtained from Chevron and used for the demulsification experiments.Infrared spectra were recorded for liquid samples neat on an Avatar 360 FT-IR spectrometer. Solution 1H and 13C NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer. Thermogravimetric analyses were performed with a Thermal Sciences STA 1500 instrument. Heats of protonation were recorded using an isothermal mixing and reaction calorimeter, model superCRC 208–110. Conductivity of surfactant solutions was measured using a JENWAY conductivity meter 4071. Height measurements for crude oil demulsifying experiments were made with an Eberbach cathetometer. The synthesis of the amphiphiles and their corresponding bicarbonate salts is described in the supporting information. Water solubility of compounds 1a–6a and the extent of conversion of each base to the bicarbonate salt by CO2 were determined by NMR (details in the ESI†). The logKow values were predicted using the ALOGPS 2.1 software, which calculates the logKow value for the given structure using nine different algorithms and then averages these values.9–11
3 Results and discussion
3.1 Synthesis of the switchable surfactants
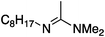
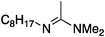
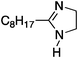
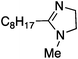
To study the influence of different head groups on the properties and performance of the switchable surfactants, a small library of bases with long alkyl chains was synthesized (Scheme 1). N′-Octyl-N,N-dimethylacetamidine was chosen to represent the alkylacetamidine structures studied previously.5 Other bases were prepared with similar alkyl chain lengths. Two cyclic imidazoline structures (2a and 3a) were prepared by reaction of the long chain carboxylic acid with ethylenediamine or N-methylethylenediamine, respectively.12 The aryl acetamidine structure 4a was synthesized in close analogy to surfactant 1a, starting from the corresponding aniline. Guanidine 5a is accessible through reaction of N,N,N′′,N′′-tetramethylchloroformamidinium chloride with octylamine.13,14 The commercially available N,N-dimethyloctylamine 6a was also included in this study.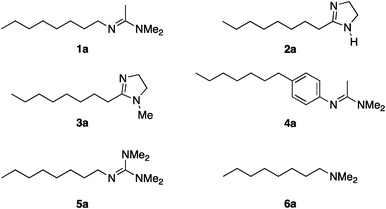 | ||
| Scheme 1 Bases evaluated in this study. | ||
3.2 Surfactant solubility
The solubilities of bases 1a–6a and octylamine in D2O under air were determined by means of NMR spectroscopy (Table 1). Furthermore, the logKow-values were calculated for all compounds using the ALOGPS 2.1 software (see experimental section).The octyl amidines 1a–3a and the octyl guanidine 5a posses a significantly higher water-solubility than 1-octylamine, suggesting that the amidine and guanidine groups are relatively hydrophilic due to their high basicity. The base with the greatest solubility is the most basic compound, guanidine 5a. This compound is so very basic (the pKa of protonated pentamethylguanidine is reported to be 15.615) that it is largely protonated in water, existing primarily as the hydroxide salt. Its high solubility is almost entirely due to the solubility of the hydroxide salt and not to the solubility of the neutral compound. The solubility of alkylamidine 1a is also increased by the formation of the hydroxide salt, but to a lesser extent. Considering a pKaH (pKa of the protonated form) of 12.1, one can calculate that at a concentration of 4.9 g L−1, 54% of the amidine is protonated. The other bases are sufficiently weak that conversion to the hydroxide salt does not greatly increase the solubility.
Comparing compounds of roughly equal basicity shows that hydrogen-bond donating ability also contributes to solubility, although not as strongly as does basicity. Thus 2a is significantly more soluble than 3a and octylamine is more soluble than 6a, presumably because of the hydrogen-bond donating ability of the N–H groups. The presence of an aromatic ring adjacent to the amidine group greatly decreases the solubility of the amphiphiles, as the arylamidine 4a shows particularly low solubility in D2O.
The logKow data were calculated for the neutral compounds and therefore do not take into account the formation of hydroxide salts. Thus, these data show that 5a and 1a are more hydrophobic than their aqueous solubilities would suggest.
| # | Molecule | Solubility in D2O (g L−1) | Predicted logKow |
|---|---|---|---|
| - |
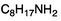
|
1.40 ± 0.12 | 2.9 |
| 1a |
|
4.90 ± 0.08 | 4.1 |
| 2a |
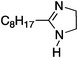
|
5.10 ± 0.45 | 3.5 |
| 3a |
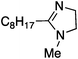
|
1.70 ± 0.02 | 3.8 |
| 4a |
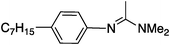
|
0.10 ± 0.06 | 5.5 |
| 5a |
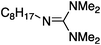
|
39.0 ± 4.0 | 3.9 |
| 6a |
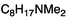
|
0.60 ± 0.05 | 3.6 |
3.3 Basicity
Next, the basicity of the compounds was examined (Table 2). We compared the pKaH values of the amphiphiles or closely related structural analogues. The pKaH data for guanidines is rather controversial: N,N,N′,N′,N′′-pentamethylguanidine, the closest analogue to compound 5a for which literature data is available, is reported to have a pKaH of either 13.816 or 15.6.15 Regardless of the true pKaH, pentaalkylguanidines are much more basic than any of the other bases in the present study. In addition, the heats of protonation of the bases were determined by calorimetry and are comparable to literature examples of structural analogues (88 kJ mol−1 for 1,1,3,3-tetramethylguanidine).17 According to the pKaH values, the basicities of the compounds decrease in the order:guanidine 5a ≫ alkylamidine 1a > imidazolines 2,3a > N′-arylamidine 4a > 3° amine 6a
| # | Molecule | −ΔHrxn, kJ mol−1 | pKaH (H2O scale) |
|---|---|---|---|
| a Value measured in H2O. b Value for 2-ethyl imidazoline.19 c Value for 1,12-bis(1-methyl-4,5-dihydro-1H-imidazol-2-yl)dodecane (an N-methylalkylimidazoline).20 d Value for the p-methyl analog.21 e Values for N,N,N′,N′,N′′-pentamethylguanidine.15,16 f Value for dimethylhexylamine.22 | |||
| 1a |
|
71 ± 1 | 12.2a |
| 2a |
|
47 ± 0.8 | 11.1b |
| 3a |
|
54 ± 0.7 | 11.0c |
| 4a |
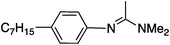
|
48 ± 0.3 | 10.8d |
| 5a |
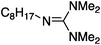
|
83 ± 0.2 | 13.8 or 15.6e |
| 6a |
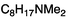
|
53 ± 0.5 | 10.0f |
The protonation enthalpies suggest almost the same sequence, except that there is very little difference between the basicities of the imidazolines, the amine, and the N′-arylacetamidine by this measure. By either measure, the aryl compound 4a is significantly less basic than the alkyl acetamidine 1a; a result which arises from the incorporation of the electron-withdrawing phenyl group onto the acetamidine head group.18 The trend in basicity is almost identical to the trend in the solubilities in water, again suggesting that basicity is the deciding factor in determining the solubility in water when other factors (molecular weight and hydrogen-bond donating ability) are roughly equal.
3.4 CO2-triggered switching
The ability of the bases to undergo a reversible CO2-triggered transition was investigated using NMR spectroscopy (eqn (2), where B is a basic functional group).| R–B + CO2 + H2O ⇌ R–BH+ + HCO3− | (2) |
Reaction of these bases with CO2 and water in DMSO-d6 causes changes in the 1H and 13C{1H} NMR spectra consistent with protonation of the base and the formation of a bicarbonate anion. In the 13C{1H} NMR spectrum, a new peak between 159.3 and 159.9 ppm represents the bicarbonate anion (in contrast, a carbonate anion would appear at 168 ppm and a mix of bicarbonate and carbonate ions would appear at 162–165 ppm).23 For the acetamidines (1a, 4a), the central carbon of the amidine group, which typically appears at 157–159 ppm in the absence of CO2, is shifted to 162–164 ppm, indicative of protonation at the adjacent nitrogen. The addition of hydrochloric, acetic, or triflic acid to the acetamidines causes an identical shift in the position of the central carbon atom's NMR signal.
Further confirmation of the products was obtained by repeating the reaction of the bases with CO2 and water in CH2Cl2. CO2 was bubbled through the solution until the solvent was almost completely driven off. The IR spectra of the resulting pastes each showed a new signal at 832–834 cm−1, which corresponds to the out-of-plane bending vibration frequency of the bicarbonate anion.24,25 In all cases, no signal at 880 cm−1 could be observed, which would correspond to the out-of-plane bending vibration frequency of the carbonate anion. In addition, other characteristic bicarbonate vibrations were observed, such as the C–OH bend (∼1000 cm−1) and C–OH stretch (∼1350 cm−1). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretch vibration overlaps with the signal of the CN vibration of the amidines or guanidines and so it is only observed in the case of the tertiary amine 6b.
O asymmetric stretch vibration overlaps with the signal of the CN vibration of the amidines or guanidines and so it is only observed in the case of the tertiary amine 6b.
The conversion of the bases to the bicarbonate species upon purging with CO2 was studied by 1H NMR spectroscopy in DMSO-d6 in the presence of H2O. The percent protonation was determined by comparing the chemical shifts of the protons on the carbon adjacent to the nitrogen atoms of a surfactant treated with CO2 with those of a fully protonated species of the same surfactant. Conversion to the bicarbonate salt in DMSO was found to be highly dependent on the amount of water added (Table 3). Because high conversions were observed for all surfactants when 200 μl of water was used (≥ 89%), the experiments were then performed with only 25 μl water present. The conversions at this water concentration were lower and spread over a much greater range, allowing comparisons of the different bases. Conversion data were not obtained for base 4a, due to its poor solubility in DMSO. No protonation occurred in the absence of CO2, even when the very basic guanidine 5a was used. A comparison of the conversion in wet DMSO with the aqueous scale pKaH shows that the minimum pKaH needed to achieve a conversion of > 90% is 10 at high H2O concentration and 13 at low H2O concentration (Fig. 1). While almost complete conversion (97%) was observed for the most basic surfactant 5a in water-poor conditions, only 68% conversion was observed for amidine 1a, which is less basic. Both imidazolines 2a and 3a had roughly 20% conversion; the presence or absence of an N–H bond in the neutral compound apparently makes little difference. Tertiary amine 6a, although it has a similar protonation enthalpy to the imidazolines, had almost no conversion to the bicarbonate salt in the presence of 25 μl of water. Nevertheless, the tertiary amines could prove to be useful (and inexpensive) switchable surfactants because the conversion is high when the water concentration is high.
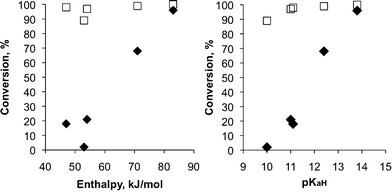 | ||
| Fig. 1 Effect of enthalpy of protonation (left) and pKaH (right) on the conversion of bases 1a, 2a, 3a, 5a, and 6a to the bicarbonate salt in a mixture of 0.6 mL of DMSO and either 25 μL (◆) or 200 μL (□) of water under CO2 at 40 °C. | ||
| # | Molecule | Water, μL | T/°C | Conversion (%) |
|---|---|---|---|---|
| a Conditions: 0.11 mmol of base, 0.6 ml of DMSO-d6. | ||||
| 1a |
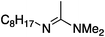
|
25 | 40 | 68 |
| 50 | 40 | 91 | ||
| 200 | 40 | 99 | ||
| 25 | 60 | 28 | ||
| 2a |
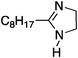
|
25 | 40 | 18 |
| 200 | 40 | 98 | ||
| 3a |
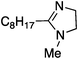
|
25 | 40 | 21 |
| 200 | 40 | 97 | ||
| 5a |
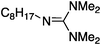
|
25 | 40 | 96 |
| 25 | 60 | 97 | ||
| 6a |
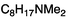
|
25 | 40 | 2 |
| 200 | 40 | 89 | ||
Because compounds 1a and 5a had the greatest conversion to the protonated form, their reactivity was investigated at 60 °C to see how the temperature affected the equilibrium position. Compound 5a was the most basic of all compounds tested and even at 60 °C was almost quantitatively converted to the guanidinium bicarbonate form. Higher temperatures would be required to convert the guanidinium bicarbonate species back to compound 5a, which would limit its application as a useful switchable surfactant. Compound 1a, being somewhat less basic, showed a much lower conversion at 60 °C. However, it is important to note that this is the conversion in DMSO; the conversion to the protonated form would be greater in pure water.
Conductivity measurements were undertaken to monitor the switching process from the uncharged base to the bicarbonate salt upon addition of CO2. Initial experiments of this type were performed in an EtOH–H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1 v/v) mixture due to the low solubility of the bases in neat H2O. For compounds 1a, 3a, and 6a, addition of CO2 caused the expected transformation of a low conductivity solution (neutral amine/amidine) to a high conductivity solution (bicarbonate salt). Removal of CO2 by bubbling argon gas led to a drop in the conductivity in the case of the tertiary amine 6a (entry 11 of Table 4 and Fig. 2a). The decrease was slower when amidines 1a and 3a were used, due to the higher basicity of these two compounds (Fig. 2a). In contrast to these results, a similar experiment with the much more basic guanidine 5a started with a very high initial conductivity due to the protonation of the base by H2O and the addition of CO2 lead to a decrease in conductivity. This somewhat counter-intuitive observation is attributed to the guanidine existing in the solution largely as the guanidinium hydroxide salt and, upon CO2 addition, conversion of that salt to the corresponding bicarbonate. That conversion lowers the conductivity because most anions in water have lower ion mobility than hydroxide. The ionic mobility of OH− in water is about 5 times that of larger anions such as acetate26 (no literature data were found for bicarbonate).
:1 or 2:1 v/v) mixture due to the low solubility of the bases in neat H2O. For compounds 1a, 3a, and 6a, addition of CO2 caused the expected transformation of a low conductivity solution (neutral amine/amidine) to a high conductivity solution (bicarbonate salt). Removal of CO2 by bubbling argon gas led to a drop in the conductivity in the case of the tertiary amine 6a (entry 11 of Table 4 and Fig. 2a). The decrease was slower when amidines 1a and 3a were used, due to the higher basicity of these two compounds (Fig. 2a). In contrast to these results, a similar experiment with the much more basic guanidine 5a started with a very high initial conductivity due to the protonation of the base by H2O and the addition of CO2 lead to a decrease in conductivity. This somewhat counter-intuitive observation is attributed to the guanidine existing in the solution largely as the guanidinium hydroxide salt and, upon CO2 addition, conversion of that salt to the corresponding bicarbonate. That conversion lowers the conductivity because most anions in water have lower ion mobility than hydroxide. The ionic mobility of OH− in water is about 5 times that of larger anions such as acetate26 (no literature data were found for bicarbonate).
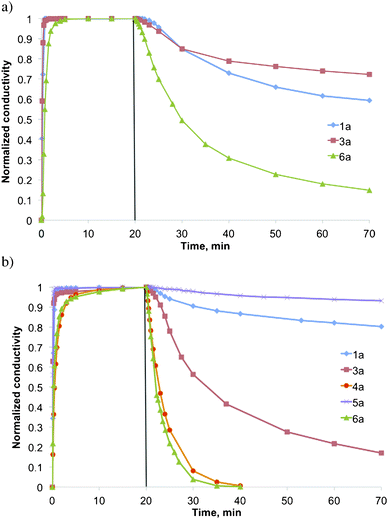 | ||
| Fig. 2 Normalized conductivity of solutions of selected bases in (a) EtOH–water (3:1 mixture) or (b) anhydrous EtOH during bubbling of CO2 for 20 min and then argon through the solutions at room temperature. To facilitate the comparison of the data, the normalized conductivity of each base was determined by setting the initial conductivity (C0) of each compound to 0 and the conductivity after 20 min of CO2 treatment to 1. | ||
| Entry | Base | Solvent | C0/μS/cm | C(CO2)/μS/cma | C(Ar)/μS/cma |
|---|---|---|---|---|---|
| a Reading taken after 20 min of CO2 or Ar treatment at room temperature. | |||||
| 1 | 1a | EtOH | 9 | 330 | 289 |
| 2 | 1a | EtOH–H2O (3:1) |
106 | 956 | 602 |
| 3 | 3a | EtOH | 39 | 363 | 160 |
| 4 | 3a | EtOH–H2O (3:1) |
76 | 690 | 507 |
| 5 | 4a | EtOH | 4 | 148 | 5 |
| 6 | 5a | EtOH | 79 | 404 | 390 |
| 7 | 5a | EtOH–H2O (1:1) |
1269 | 470 | - |
| 8 | 5a | DMSO–H2O (1:1) |
489 | 324 | - |
| 9 | 5a | DMSO | 13 | 273 | 114 |
| 10 | 6a | EtOH | 6 | 69 | 7 |
| 11 | 6a | EtOH–H2O (3:1) |
7 | 481 | 149 |
Additional conductivity experiments to further compare the switching of the different bases were performed in neat anhydrous ethanol, with the expectation that the lower solution polarity and the weaker acidity of ethanol would decrease the extent of guanidine protonation in the absence of CO2. Indeed, in this medium, the solution of guanidine had a low initial conductivity that rose significantly upon CO2 addition. The conductivities of all base solutions increased significantly upon CO2 addition, from a 5-fold increase for guanidine 5a to 37-fold increase for 1a and 4a (Table 4).The normalized conductivities of bases 1a, 3a, 4a, 5a, and 6a are depicted in Fig. 2b. As expected, the basicity of the surfactant head group has a major influence on the switching process. While the more basic surfactants, guanidine 5a and the amidines 1a and 3a, only need bubbling of CO2 for less than a min at room temperature for the conductivity to reach more than 96% of its maximum, the less basic surfactants 4a, and 6a need a longer treatment. However, all surfactants can be switched to exhibit their highest conductivity after 20 min of CO2 treatment. The biggest difference between the surfactants becomes apparent when Ar bubbling is used to displace the CO2 in order to deactivate the surfactants (by converting them back to the neutral bases). Solutions of compounds 4a, and 6a can be switched back to their starting conductivity within 20 min of Ar treatment at room temperature. Surfactants 1a, 3a and 5a on the other hand still remain partially protonated with 76% (1a), 44% (3a) and 93% (5a) of their maximum conductivity after 50 min of purging with Ar. Increasing the temperature to 45 °C increases the deactivation rate, however even then the conductivity of the guanidine surfactant 5a only drops to 79% of the maximum conductivity after 50 min.
Finally, surfactants 4a and 6a were submitted to three consecutive cycles of CO2 and then argon treatment. In both cases good consistency over all three cycles was observed. As a representative example, Fig. 3 shows three complete cycles for surfactant 4a.
 | ||
| Fig. 3 Conductivity of surfactant 4a in ethanol in the presence and absence of CO2 at room temperature. Three consecutive cycles are shown. | ||
To determine if the addition of CO2 to compounds 1a–6a leads to a significant increase in their emulsion-stabilizing ability, the stabilities of water–decane emulsions containing different bases in the presence or absence of CO2 were examined. As Fig. 4 shows, CO2 plays a crucial role for the stability of these emulsions. In the presence of CO2, emulsions with the more basic compounds 1a, 3a, 4a, or 5a stabilize emulsions for up to 48 h, while without CO2 the emulsions start breaking within a few minutes in all cases. In contrast, the less basic dimethyloctylamine 6a is not able to stabilize the emulsion with or without CO2 present. Compound 2a, the only solid base, was not studied because it formed a solid dispersion in the water–decane mixture.
 | ||
| Fig. 4 Emulsion stability test using surfactant 1 (a), 3 (b), 4 (c) and 5 (d) under air (left vial) or CO2 (right vial) after 2 min (left column) and 24 h (right column). | ||
3.5 Demulsifying ability
The ability of these bases to demulsify 2:1 v/v crude oil–water emulsions was evaluated; the experimental procedure used was similar to that previously described,5 except North Sea oil, a somewhat heavier crude oil, was used. A mixture containing the base, oil and water was agitated and then allowed to settle on the bench top at room temperature under air. The separation was monitored as a function of time by height measurements of the oil–emulsion and emulsion–water interfaces using a cathetometer. The emulsions showed visible separation including creaming (formation of an oil layer on top of the emulsion) and sedimentation (formation of a water layer below the emulsion), but the rate and degree of separation depended upon the choice of the base.
An emulsion prepared in this way without base showed no visible separation during the first three days of observation. Sedimentation started on the fourth day; at the end of the day the produced water layer was completely clear and no emulsion remained.
Emulsions containing compounds 1a, 2a, and 4a showed sedimentation, while those containing compounds 2a, 3a, and 4a showed creaming, and those containing compounds 5a, 6a showed no separation at all in the first 24 h. The emulsion containing alkyl acetamidine 1a showed the earliest initial water separation, within the first 2 h of settling, although the aqueous layer remained partially cloudy until 4 days had elapsed when the original volume of water was fully separated and was clear. The height of the boundary between the water and emulsion layers is shown as a function of time in Fig. 5a.
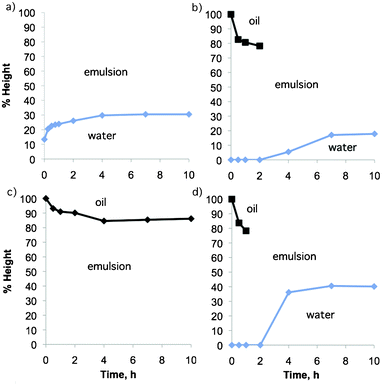 | ||
| Fig. 5 Height of phases observed during the demulsification of an emulsion of 4 ml crude oil, 2 ml water and 0.29 mmol of (a) 1a, (b) 2a, (c) 3a, (d) 4a at room temperature. No separation was observed in the first 24 h with compounds 5a and 6a. | ||
Imidazoline 2a began to cause separation of water from the emulsion after 2 h but it took 4 days to cause full separation of the oil and aqueous layers and the water was clear (Fig. 5b). Creaming occurred initially but after 2 h it became visually difficult to see the boundary between the emulsion and oil layers. Employing the other imidazoline, 3a, resulted in no sedimentation, even after 7 days of monitoring the emulsion. It did, however, show some creaming in the first day (Fig. 5c). The reason for the difference in performance is unknown.
Aryl acetamidine 4a was the first compound to cause complete demulsification, giving a clear water phase after only 4 h (Fig. 5d).
Bases 1a, 2a, 4a, and 6a began to cause water separation before the blank sample (i.e. < 3 days). However, only 4a and 6a were able to achieve full demulsification more quickly than the blank (i.e. < 4 days). Bases 3a and 5a did not promote complete demulsification of the crude oil–water emulsion even after 7 days. Compound 4a was the only base to cause complete separation of the oil and water within 2 days.
The demulsification studies of these crude oil–water emulsions showed that the demulsifying ability of the base is strongly dependent upon the structure of the head group. Not all of the compounds tested were able to accelerate the separation of an oil and water emulsion; compound 4a was the only one able to do so more effectively than the alkyl acetamidine 1a (Fig. 6). There was no direct correlation between the demulsifying capability and the basicity of the compounds.
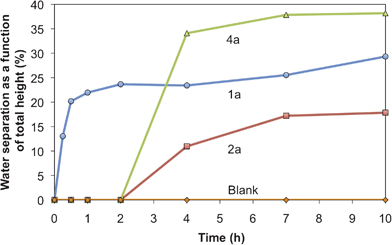 | ||
| Fig. 6 Sedimentation over time of emulsions of crude oil (4 mL) and water (2 mL) with 1a, 2a, or 4a (0.29 mmol) in comparison to a blank sample (containing no switchable surfactant) over 10 h at room temperature. | ||
4 Conclusions
We were able to show that the nature of the nitrogen-containing head group of long-alkyl chain molecules has a major influence on their performance as CO2-switchable surfactants. While the alkylacetamidine, pentaalkylguanidine, 2-alkykimidazolines, arylacetamidine, and trialkylamine compounds can all be easily protonated with carbonic acid in the presence of a large amount of water, only the more basic alkyl guanidine and alkyl amidine compounds show a high conversion to the protonated, more surface-active species in organic solvent at low water concentrations. The different basicities of the compounds strongly influence the switching rates of the switchable surfactants.All but the tertiary amine demonstrated the ability to stabilize decane–water emulsions of an organic solvent and water. The arylacetamidine switchable surfactant shows particular promise; it not only stabilized emulsions but was also capable of being “switched off” (converted back to the neutral form) far more quickly than the other amidines and the guanidine. Rapid switching off, especially at room temperature, would be of significant practical advantage. The tertiary amine is also switched off rapidly. Despite the failure of the tertiary amine in the decane–water emulsion tests, we have successfully performed emulsion polymerization using a tertiary amine bicarbonate salt switchable surfactant.27
While the four amidines (two acyclic and two cyclic) showed demulsifying ability for crude oil–water mixtures, the two acetamidines were superior to the imidazolines.
Thus, the design of the head group and, in particular the basicity of the head group, needs to be carefully considered and tailored to meet the needs of the specific application.
Acknowledgements
The authors gratefully acknowledge the financial support of the Natural Sciences and Engineering Council of Canada, Chevron Energy Technology Company, and the Canada Research Chairs program for funding, and Dr Don Kuehne and Dr Cesar Ovalles for valuable discussions.References
- T. Mezger, O. Nuyken, K. Meindl and A. Wokaun, Prog. Org. Coat., 1996, 29, 147–157 CrossRef CAS.
- A. Lottermoser, Trans. Faraday Soc., 1934, 31, 200–204 RSC.
- E. R. Moore and N. A. Lefevre, U. S. Patent Pat., 4,623,678, 1986 Search PubMed.
- C. I. Fowler, C. Muchemu, R. E. Miller, L. Phan, M. F. Cunningham and P. G. Jessop, Macromolecules, 2011, 44, 2501–2509 CrossRef CAS.
- Y. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958–960 CrossRef CAS.
- T. Arthur, J. R. Harjani, L. Phan, P. G. Jessop and P. V. Hodson, Green Chem., 2012, 14, 357–362 RSC.
- C. Liang, J. R. Harjani, T. Robert, E. Rogel, D. Kuehne, C. Ovalles, V. Sampath and P. G. Jessop, Energy Fuels, 2012, 26, 488–494 CrossRef CAS.
- Y. Qin, H. Yang, J. Ji, S. Yao, Y. Kong and Y. Wang, Tenside Surf. Det., 2009, 46, 294–296 CAS.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS.
- I. V. Tetko and V. Y. Tanchuk, J. Chem. Inf. Model., 2002, 42, 1136–1145 CrossRef CAS.
- VCCLAB, Virtual Computational Chemistry Laboratory, http://www.vcclab.org, accessed Nov ‘09.
- Z. Shi and H. Gu, Synth. Commun., 1997, 27, 2701–2707 CrossRef CAS.
- T. Fujisawa, K. Tajima and T. Sato, Bull. Chem. Soc. Jpn., 1983, 56, 3529–3530 CrossRef CAS.
- G. Wieland and G. Simchen, Liebigs Ann. Chem., 1985, 2178–2193 CrossRef CAS.
- D. Margetic, in Superbases for Organic Synthesis, ed. T. Ishikawa, Wiley, Chichester, UK, 2009 Search PubMed.
- S. J. Angyal and W. K. Warburton, J. Chem. Soc., 1951, 2492–2494 RSC.
- K. Izutsu, T. Nakamura, K. Takizawa and A. Takeda, Bull. Chem. Soc. Jpn., 1985, 58, 455–458 CrossRef CAS.
- J. Oszczapowicz and E. Raczynska, J. Chem. Soc., Perkin Trans. 2, 1984, 1643–1646 RSC.
- J. Elguero, E. Gonzalez, J.-L. Imbach and R. Jacquier, Bull. Soc. Chim. Fr., 1969, 4075–4077 CAS.
- M. l. Calas, M. Ouattara, G. Piquet, Z. Ziora, Y. Bordat, M. L. Ancelin, R. Escale and H. Vial, J. Med. Chem., 2007, 50, 6307–6315 CrossRef CAS.
- M. J. Cook, A. R. Katritzky and S. Nadji, J. Chem. Soc., Perkin Trans. 2, 1976, 211–214 RSC.
- B. A. Lur'e and V. M. Kozhukhova, J. Gen. Chem. USSR (Engl. Transl.), 1991, 61, 2726–2730 CAS , 2532–2535.
- F. Mani, M. Peruzzini and P. Stoppioni, Green Chem., 2006, 8, 995–1000 RSC.
- D. L. Bernitt, K. O. Hartman and I. C. Hisatsune, J. Chem. Phys., 1965, 42, 3553–3558 CrossRef CAS.
- A. R. Davis and B. G. Oliver, J. Solution Chem., 1972, 1, 329–339 CrossRef CAS.
- P. W. Atkins, Physical Chemistry, 2nd edn, W. H. Freeman, San Francisco, 1982 Search PubMed.
- C. I. Fowler, P. G. Jessop and M. F. Cunningham, Macromolecules, 2012, 45, 2955–2962 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01242a |
| This journal is © The Royal Society of Chemistry 2012 |