Microwave assisted isomerization of alkenyl aromatics over solid base catalysts: an understanding through theoretical study†
C. M.
Jinesh
a,
Anik
Sen
b,
Bishwajit
Ganguly
*b and
S.
Kannan
*a
aDiscipline of Inorganic Materials & Catalysis, Central Salt and Marine Chemicals Research Institute, Council of Scientific and Industrial Research (CSIR), GB Marg, Bhavnagar 364 002, India. E-mail: skannan@csmcri.org; kanhem1@yahoo.com; Fax: +91-278-2567562; Tel: +91-278-2567760 Ext.703
bAnalytical Science Discipline, Central Salt and Marine Chemicals Research Institute, Council of Scientific and Industrial Research (CSIR), GB Marg, Bhavnagar 364 002, India. E-mail: ganguly@csmcri.org; Tel: +91-278-2567760 Ext. 677
First published on 21st May 2012
Abstract
Microwave assisted isomerization of estragole to anethole was studied over MgAl and NiAl layered double hydroxides with different M(II)/Al atomic ratios as solid base catalysts. Reactions under microwave overcame the challenges like higher reaction temperature, longer time, larger solvent volume and unproductive recyclability that were encountered with conventional thermal heating for this reaction. MgAl4 (catalyst with Mg/Al atomic ratio of 4.0) gave maximum conversion of 99% with a substrate to catalyst weight ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 140 °C in 90 min using 4 ml DMF. A good correlation was obtained between the activity and Brønsted basicity derived using Hammett studies. Solvent with high polarity and high boiling point assisted the reaction and the catalyst was reusable for up to six cycles without significant loss in activity. Studies extended for different alkenyl aromatics under optimized conditions over MgAl4 revealed a very high conversion (>97%) for estragole and allylbenzene (>99%) while poor conversion for eugenol (19%). The variation in isomerization activity of alkenyl aromatics was rationalized with DFT calculations. The B3LYP/6-31+G* calculated results revealed that the conversion of these alkenyl aromatics was dependent on the substituents attached to the aromatic ring and governed by the pKa of the reactive sites in such systems. Methodology reported here offers an alternate energy efficient and environmentally benign route for the synthesis of alkenyl aromatics, which are extensively used as perfumery chemicals.
:1 at 140 °C in 90 min using 4 ml DMF. A good correlation was obtained between the activity and Brønsted basicity derived using Hammett studies. Solvent with high polarity and high boiling point assisted the reaction and the catalyst was reusable for up to six cycles without significant loss in activity. Studies extended for different alkenyl aromatics under optimized conditions over MgAl4 revealed a very high conversion (>97%) for estragole and allylbenzene (>99%) while poor conversion for eugenol (19%). The variation in isomerization activity of alkenyl aromatics was rationalized with DFT calculations. The B3LYP/6-31+G* calculated results revealed that the conversion of these alkenyl aromatics was dependent on the substituents attached to the aromatic ring and governed by the pKa of the reactive sites in such systems. Methodology reported here offers an alternate energy efficient and environmentally benign route for the synthesis of alkenyl aromatics, which are extensively used as perfumery chemicals.
1. Introduction
Isomerization of alkenyl aromatics has high industrial demand as intermediates for various perfumery chemicals (Scheme 1).1a,b Among alkenyl aromatics, trans-anethole has many applications in food and beverage industry, formulation of oral sanitation products, pharmaceutical compounds, and perfumery chemicals.2a–d Anethole, also known as isoestragole occurs in nature as both cis and trans forms, wherein trans-isomer being more abundant. Anethole is a major component of several essential oils, including anise seed oil (80–90%), star anise oil (>90%) and sweet fennel oil (80%). The total global production of trans-anethole is approximately 0.75 million metric tonnes per annum. The increasing demand for anethole led to the development of new synthetic routes other than isolation from essential oils. The most viable one is the simple base catalyzed isomerization of estragole (methyl chavicol) to its propenyl derivative, anethole. Conventionally, alkalis such as KOH in alcoholic solutions (most often in higher alcohols) at high temperatures were used for isomerization of alkenyl aromatics.3a,b Kameda and Yoneda have reported homogeneous isomerization of estragole over [RhH2(Ph2N3)(PPh3)2] in dimethyl sulfoxide (DMSO) under hydrogen atmosphere (1 atm) at 30 °C.4 Very recently, isomerization of estragole to anethole was reported over Ru-complexes that gave predominantly trans selective products.5a,b Solid base catalysts explored for this class of isomerization reaction offer easy separation, better handling and avoid polluting effluent discharge.6a–c A recent approach by Al-Maskery et al. on this reaction using K2CO3 on alumina as solid base catalyst showed that the catalyst was active at low temperature (~ 30 °C). However, deactivation observed for this catalyst even at such low temperature indicated its limitation.6c We have previously reported the utility of as-synthesized layered double hydroxides (LDHs) as promising heterogeneous solid base catalysts for such double bond isomerization of alkenyl aromatics.7a–c LDHs are promising heterogeneous solid base catalysts that are explored for diverse base catalyzed transformations such as aldol condensation, Michael addition, isomerization, C–C bond coupling reactions, and Friedel–Crafts acylation to mention a few.8a–f Layered double hydroxides, otherwise known as hydrotalcite-like (HT-like) compounds, are a class of anionic clay materials with general formula [M(II)1−xM(III)x(OH)2][Ax/nn−]·mH2O, where M(II) and M(III) are divalent and trivalent cations, An− is the interlayer anion and x can generally have values between 0.2 and 0.33.9 Our earlier report on isomerization of estragole to anethole over binary hydrotalcites revealed that MgAl-LDH with Mg/Al atomic ratio of 4.0 was the most active catalyst and the conversion increased slightly upon incorporation of Ru and Cs under conventional thermal heating.10 The drawbacks under thermal heating are high reaction temperature (200 °C), use of large solvent volume (20 ml) and longer reaction time (up to 6 h). Microwave irradiation (MWI) involves dielectric heating and is simple and fast that provides significant enhancement in the reaction rates. MWI processes are proposed as more economical through reduced consumption of energy involved for a reaction as achieved in shorter reaction times.11a,b MWI has been explored for many organic transformation using hydrotalcites as catalysts.12a,b Thach and Strauss previously used a microwave batch reactor for isomerization of estragole under aqueous conditions using 0.2 M NaOH and obtained a conversion of 81% at 230 °C.13 Recently Crochet and co-workers reported 99% yield of anethole with high trans selectivity from estragole within 15 min under microwave irradiation using Ru-complexes as homogeneous catalysts.5a,b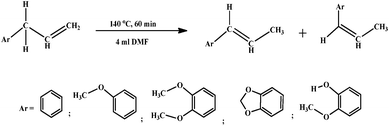 | ||
| Scheme 1 Isomerization of alkenyl aromatics. | ||
With this background, we carried out isomerization of alkenyl aromatics using a series MgAl and NiAl LDHs as heterogeneous catalysts under microwave irradiation. Our aim was to overcome the challenges of higher temperature, larger solvent volume, longer reaction time, and unproductive recyclability that were encountered with the conventional thermal heating method for this reaction. Attempts were made to correlate the observed activity with the basicity determined using Hammett studies. The DFT calculations performed with the different alkenyl aromatics revealed the reasons behind the variation in their activity over an active catalyst. To the best of our knowledge, this is the first report on microwave expedited isomerization of alkenyl aromatics over inexpensive LDH-derived solid base heterogeneous catalyst and discloses the understanding on the variation in isomerization activity of different alkenyl aromatics through theoretical studies that validates our earlier reports.
2. Experimental
2.1 Catalysts synthesis
A series of MgAl and NiAl binary hydrotalcites with carbonate as interlayer anion was prepared by low supersaturation technique whose details are given elsewhere.8f Samples thus prepared were named as M(II)M(III)x where x stands for M(II)/M(III) atomic ratio.2.2 Characterization
Elemental chemical analysis of the samples was determined using inductively coupled plasma emission spectrometry (ICP-OES; Perkin Elmer, OES, Optical 2000 DV) by digesting the sample in minimum amount of concentrated HNO3. The samples were characterized by Powder X-ray diffraction (PXRD; Rigaku-MiniFlex) system using Cu-Kα radiation (λ = 1.5406 Å). Identification of the crystalline phases was made by comparison with the JCPDF files.14 Fourier transformed-infrared (FT-IR) spectra were recorded in a Perkin-Elmer FT1730 instrument using KBr pellets; 64 spectra (recorded with a nominal resolution of 4 cm−1) were accumulated and averaged to improve the signal-to-noise ratio. Thermogravimetric (TG) analysis was carried out in a Mettler TGA/SDTA 851e and the data were processed using Stare software, in flowing nitrogen (flow rate, 50 ml min−1), at a heating rate of 10 °C min−1. Specific surface area and pore size analysis of the samples were measured by nitrogen adsorption at −196 °C using a sorptometer (ASAP-2010, Micromeritics). The samples were degassed at 80 °C for 4 h prior to measurements and the data were analyzed using published built-in software.2.3 Catalytic isomerization of alkenyl aromatics
Isomerization of alkenyl aromatics was conducted in a microwave synthesis work station (Sineo, MAS-II) equipped with direct sensing; the microwave power could be adjusted between 0–1000 W in order to achieve the desired reaction temperature. The substrate, solvent and catalyst were charged together and subjected to reaction temperature using 100% MWI. The products were analyzed by gas chromatography (Varian 450-GC) with a capillary column (Factor Four VF-1) and FID detector. Identification of the products was also further verified using GC-MS (Shimadzu QP 2010). Quantification of the products was done using tetradecane as internal standard. For reusability studies, one of the catalysts, MgAl4 was assessed wherein the used catalyst was filtered, washed with DMF and finally rinsed with acetone, dried and checked for isomerization of estragole. For conventional heating studies, isomerization was conducted in a batch reactor (50 ml), wherein the substrate, solvent and catalyst were charged together and kept in a preheated oil bath at desired reaction temperature.15 The isomerisation of the alkenyl aromatics along with the geometries are given in Scheme 1.2.4 Hammett indicator studies
In order to evaluate the Brønsted basicity associated with the hydroxyl groups, Hammett indicator measurements were carried out. 25 mg of catalyst was taken along with 2.5 ml of dry methanol and to that calculated amount of indicators with different pKa ranges were added separately and kept in a shaker for 3 h under N2 atmosphere.16 The solution was then titrated against 0.02 M benzoic acid in dry methanol to determine the basicity. The indicators used were methyl red (pKa = 5.1), neutral red (pKa = 6.8), phenolphthalein (pKa = 8.2), nile blue (pKa = 10.1), tropaeolin O (pKa = 11), 2, 4-dinitro aniline (pKa = 15).2.5 Computational methods
All calculations were performed with density functional theory (DFT) using Becke's three-parameter exchange functional with the correlation functional of Lee, Yang, and Parr (B3LYP).17a,b All species were fully optimized with the 6-31+G* basis set,18 and harmonic vibrational frequency calculations were used to confirm that the optimized structures were minima, as characterized by positive vibrational frequencies. The gas phase basicities (GB) were calculated from the energy change of the protonation reaction: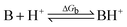
GB is defined as the negative Gibbs free energy of the reaction, GB = −ΔGb
Solvent effects were taken into account by means of the polarizable continuum model (PCM) through single-point energy calculations at the B3LYP/6-31+G* level of theory (using the gas-phase optimized geometries) with DMF as the solvent (dielectric constant, ε = 38.2).19a–e The PCM calculations, using Gaussian 03, employ the UA0 (Simple United Atom Topological Model) atomic radii when constructing the solvent cavity for the calculation of the Gibbs free energy of solvation. The pKa calculations were performed using the standard thermodynamic cycle depicted in Scheme 2.20a–e The pKa value of the acid BH+ was related to the Gibbs free energy change for the deprotonation process, which is shown below,
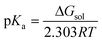
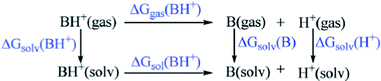 | ||
| Scheme 2 | ||
ΔGgas is the Gibbs free energy change of the reaction in the gas phase and ΔΔGsolv is the difference in solvation free energies (ΔGsolv) between products and reactants. ΔGcorr is the correction associated with the change in the standard state from the gas phase (1 atm) to solution (1 mol L−1) and its value at 298.15 K is 1.89 kcal mol−1.21 Now ΔGsol can be expressed as:
| ΔGsol = Ggas(B) + ΔGsolv(B) + Ggas(H+) + ΔGsolv(H+) − Ggas(BH)+ − ΔGsolv(BH)+ + 1.89 |
Here, the value of Gibbs free energy of the proton in the gas phase was set to −6.28 kcal mol−1 using translational entropy calculated according to the well-known Sackur–Tetrode equation22 and the value of Gibbs free energy of the proton in the DMF solvent phase was taken as −263.8 kcal mol−1.23 The ΔGsolv values in this study were determined from PCM/B3LYP/6-31+G* single-point calculations on the gas phase B3LYP/6-31+G* optimized geometry with UA0 radii and ‘scfvac’ keyword using DMF as a solvent.24a,b Both electrostatic and nonelectrostatic (i.e., cavitation, repulsion and dispersion) terms were included in the calculation of ΔGsolv values. All quantum chemical calculations were performed using the Gaussian 03 (Revision E.01) program.25
3. Results and discussion
3.1 Catalyst characterization
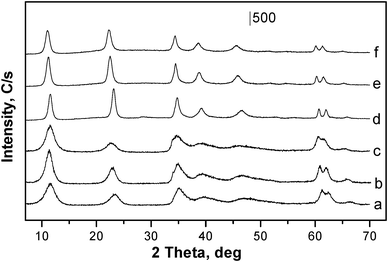 | ||
| Fig. 1 Powder X-ray diffraction patterns of: (a) NiAl2, (b) NiAl3, (c) NiAl4, (d) MgAl2, (e) MgAl3, (f) MgAl4. | ||
| Catalyst | Lattice parameters | Crystallite size (Å)a | Surface areab | Pore volumec | |
|---|---|---|---|---|---|
| a/Å | c/Å | ||||
| a Calculated using Debye–Scherrer equation considering (003) and (006) planes. b Specific surface area in m2g−1. c Pore volume in cc g−1. | |||||
| [Mg0.72Al0.28(OH)2] (CO3)0.17·0.75H2O | 3.049 | 23.03 | 56 | 97 | 0.40 |
| [Mg0.78Al0.22(OH)2] (CO3)0.11·0.86H2O | 3.068 | 23.75 | 62 | 94 | 0.68 |
| [Mg0.80Al0.20(OH)2] (CO3)0.10·0.67H2O | 3.075 | 24.00 | 79 | 91 | 0.41 |
| [Ni0.70Al0.30(OH)2] (CO3)0.15·0.75H2O | 3.021 | 22.94 | 57 | 152 | 0.33 |
| [Ni0.75Al0.25(OH)2] (CO3)0.13·0.73H2O | 3.040 | 23.22 | 53 | 146 | 0.42 |
| [Ni0.82Al0.18(OH)2] (CO3)0.09·0.52H2O | 3.053 | 23.28 | 57 | 126 | 0.36 |
This was further confirmed through FT-IR whose spectra are given in the ESI, Fig. S1.† The main band recorded around 3500 cm−1 was due to νOH stretching of hydroxyl groups from the layers and interlayer water molecule. The weak band observed at 1630 cm−1 was due to δH2O mode of interlayer water molecules and strong band at 1363 cm−1 was attributed to ν3 asymmetric stretching of carbonate vibrations shifted from its position in free carbonate species (≈1450 cm−1). It was found that the νOH stretching band slightly shifted to higher values with an increase in Mg concentration indicates stronger hydrogen bonding between the hydroxyl groups. Thermal analysis of the samples showed two well-defined weight losses as evidenced from the Fig. S2 (ESI†). The first weight loss T1 °C at around 150–180 °C is due to removal of the water molecules present in the interlayer while the second weight loss T2 °C at around 360–375 °C is ascribed to the dehydroxylation and decarbonation from hydrotalcite network. A decrease in the transformation temperatures were observed with an increase in M(II)/Al atomic ratio. This could be due to the decrease in the electrostatic interaction owing to the decrease in Al3+ content that corroborates well with the increase in the lattice parameter ‘c’ that signifies the interlayer distance. BET measurements revealed (Table 1) M(II)/Al atomic ratio did not have significant influence on the specific surface area although it was higher for Ni-containing samples compared to Mg-containing samples.
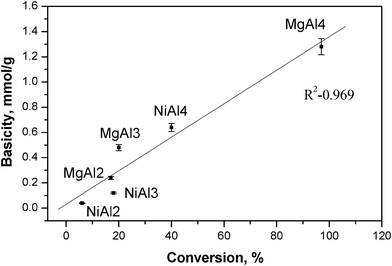 | ||
| Fig. 2 Hammett basicity-activity relationships. Error bar for basicity values measured are around 5%. | ||
3.2 Catalytic studies
| Catalysta | Basic strength | Basicity, mmol g−1c | Conversion (%) | Selectivity (%) | |
|---|---|---|---|---|---|
| Cis | Trans | ||||
| a Substrate: Estragole (200 mg); catalyst weight: 100 mg; solvent, DMF (4 ml); reaction time/temp: 60 min/140 °C; power: 300 W. b After six cycles. c Basicity was calculated on the basis of the endpoint of the titration. | |||||
| MgAl2 | 11 < H_ < 15 | 0.24 | 17 | 17 | 83 |
| MgAl3 | 11 < H_ < 15 | 0.48 | 20 | 16 | 84 |
| MgAl4 | 11 < H_ < 15 | 1.28 | 97 | 17 | 83 |
| NiAl2 | 11 < H_ < 15 | 0.04 | 6 | 10 | 90 |
| NiAl3 | 11 < H_ < 15 | 0.12 | 18 | 18 | 82 |
| NiAl4 | 11 < H_ < 15 | 0.64 | 40 | 17 | 83 |
| MgAl4b | 11 < H_ < 15 | 0.48 | 47 | 13 | 87 |
MgAl4 showed a maximum basicity of 1.28 mmol g−1 among the catalysts studied and gave maximum isomerization activity (Table 2). MgAl2 and MgAl3 showed basicity values of 0.24 and 0.48 mmol g−1 respectively. In the case of NiAl samples, NiAl4 gave maximum basicity value of 0.64 mmol g−1 and in turn showed highest activity among NiAl samples. A good correlation was observed between basicity and activity wherein activity increased with an increase in the basic strength as illustrated in Fig. 2. The trend was in good agreement with the literature wherein with an increase in Mg and Ni content the basicity as well as activity increased for hydrotalcites.8f,27
:trans ratio 14:86 at 140 °C. Influence of reaction temperature, substrate to catalyst weight ratio, and solvent was studied for the active catalyst, MgAl4, to optimize the reaction conditions. In the temperature range 80–140 °C the results showed an increase in the conversion with the temperature (Fig. 3). A maximum conversion of 94% with cis:trans ratio 13:87 was obtained at 140 °C in 1 h. A further increase in temperature (150 °C) was not achieved under MWI (at 300 W) with DMF as solvent. The results obtained here are promising and encouraging as more than 90% conversion was achieved at relatively lesser temperature and time in comparison with conventional heating (200 °C, 97% conversion in 6 h).10
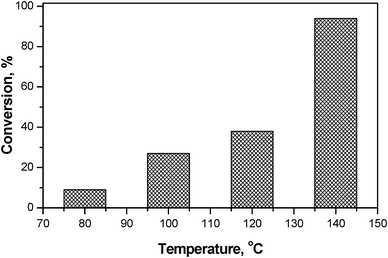 | ||
| Fig. 3 Influence of reaction temperature on conversion for MgAl4 (conditions similar to Table 2 except for temperature). | ||
Similar conditions at 140 °C through thermal heating for 1 h showed only 6% conversion (94% under microwave). The substrate to catalyst weight ratio variation (Fig. 4) revealed that the conversion increased with an increase in the weight of catalyst (i.e., decrease in substrate:catalyst weight ratio). A maximum conversion of 97% in 1 h was observed with a substrate:catalyst weight ratio of 2:1 with a cis:trans ratio of 17:83 at 140 °C.
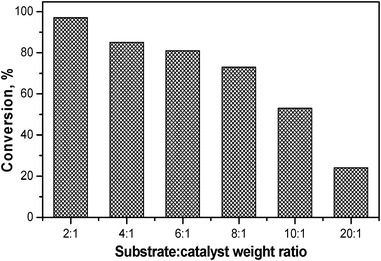 | ||
| Fig. 4 Effect of substrate: catalyst weight ratio on conversion for MgAl4 (conditions similar to Table 2 except for substrate amount). | ||
Further, the activity decreased with an increase in the volume of solvent; with 16 ml of DMF only 46% conversion was observed while under identical conditions, 97% conversion was observed using 4 ml of solvent. However, on further decrease in the volume of solvent (2 ml), the reaction temperature was not achieved. The isomerization activity of MgAl4 in different solvents was also studied whose results are given in Table 3. The solvents were categorized as high-boiling-high-polar, high-boiling-low-polar, low-boiling-high-polar and low-boiling-low-polar. The results revealed that only those solvents with high polarity and relatively high boiling point favored the reaction as it was well known that influence of MWI depends on the polarity/dielectric of the medium. High-boiling-high-polar solvents like DMF, DMSO, and dimethyl acetamide (DMA) gave good conversion of 95 ± 3% in 1 h, while all other solvents failed in giving good conversion.
| Solventa | Dielectric constant | Boiling point, °C | Conversion (%) | Selectivity (%) | |
|---|---|---|---|---|---|
| Cis | Trans | ||||
| a Substrate: Estragole (200 mg); catalyst weight: 100 mg; solvent (4 ml); reaction time/temp: 60 min/140 °C; power: 300 W. b Vapor loss observed. c Reaction temperature not attained. | |||||
| DMSO | 47.2 | 189 | 93 | 19 | 81 |
| DMF | 38.2 | 152 | 97 | 17 | 83 |
| DMA | 37.8 | 166 | 99 | 14 | 86 |
| Nitrobenzene | 34.8 | 211 | 12 | 13 | 87 |
| Acetonitrile | 36.6 | 82 | 1 | 0 | 0 |
| Methanolb | 33 | 65 | 1 | 0 | 100 |
| Butanol | 18 | 118 | 0 | 0 | 0 |
| Heptanolc | 6.7 | 178 | 33 | 17 | 83 |
| Dodecanolc | 6.5 | 259 | 11 | 12 | 88 |
| Ethanediolc | 6.9 | 197 | 0 | 0 | 0 |
| Waterb | 80 | 100 | 0 | 0 | 0 |
Thus high conversion of estragole to anethole was obtained in shorter reaction time, lower temperature and by using a reduced volume of solvent under MWI. To check the reusability of the active catalyst, MgAl4, recycling studies were done for up to six cycles (Fig. 5). Stable activity of around 95% conversion was noted for the first two cycles. A continuous although marginal decrease in the conversion was noted with the further increase in the number of cycles; the catalyst exhibited 47% conversion for the sixth cycle. It was found from Hammett study that MgAl4 after six cycles showed basicity of 0.48 mmol g−1 (Table 2). The decrease in the basicity value (from 1.28 mmol g−1 for fresh catalyst to 0.48 mmol g−1 after sixth cycle) correlated well with the activity.
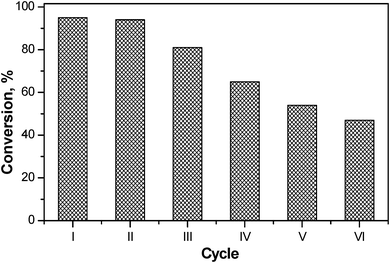 | ||
| Fig. 5 Recycle studies for MgAl4 for isomerization of estragole (conditions similar to Table 2). | ||
| Substratesa | Conversion (%) | pKa at 140 °C for H1 | pKa at 140 °C for H1a |
|---|---|---|---|
| a Substrate: 200 mg; catalyst weight: 100 mg; solvent (4 ml); reaction time/temp: 60 min/140 °C; power: 300 W. | |||
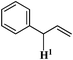 Allylbenzene Allylbenzene |
>99 | 26.28 | — |
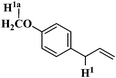 Estragole Estragole |
97 | 27.58 | 41.18 |
 Allylveratrole Allylveratrole |
88 | 26.25 | 40.70 |
 Safrole Safrole |
64 | 26.98 | 35.38 |
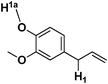 Eugenol Eugenol |
19 | 27.84 | 13.43 |
Among the alkenyl aromatics studied, allylbenzene showed maximum conversion to β-methyl styrene (>99%) while eugenol showed minimum conversion to isoeugenol (19%). Allylveratrole gave 88% conversion and safrole exhibited 64% conversion. The variation in the activity of different alkenyl aromatics could be due to variation in the adsorption potential or mode of adsorption of the substrates on the surface of the catalyst in turn due to the variation in the substituents attached to the aromatic ring. It must be mentioned here that a similar activity trend was observed under conventional thermal heating for NiAl4.7b
The difference in the activity is generally governed by the molecular dimension, functionality and mode of adsorption of the substrate on the surface. It is known that the acidity of the methylene proton (–CH2–) of allylic moiety is the key parameter and its abstraction by the basic site is critical for the double bond migration.7b In order to confirm the trend under conventional conditions, isomerization of alkenyl aromatics was assessed under similar conditions (140 °C for 6 h with substrate to catalyst ratio 2:1 with 4 ml DMF as solvent). Allylbenzene and estragole showed a conversion of 45% and 11% respectively while allylveratrole and eugenol did not show conversion. This confirms that at lower temperature thermal heating the energy needed for the reaction was not attained.
To discern the factors responsible for the observed difference in the isomerization activities of the alkenyl aromatics, DFT B3LYP/6-31+G* level calculations were performed. The isomerization reaction could be perceived to occur through the deprotonation of the allyl proton H1 by the hydroxyl group of MgAl4 catalyst.8f The B3LYP/6-31+G* calculated pKa for the allyl protons of the alkenyl aromatics at the experimental temperature of 140 °C, are given in Table 4. The calculated pKa results indicated that the extent of conversion towards the isomerization of all the studied alkenyl aromatics should be similar. However, the observed isomerization activities of these alkenyl aromatics i.e., allylbenzene to eugenol varied significantly. It was then perceived that the substituted alkenyl aromatics (estragole to eugenol) have also other deprotonation sites, which can interfere with the abstraction of allyl proton H1.
The pKa calculations performed with the deprotonation (H1a) of the substituents attached to the aromatic rings are given in Table 4. The calculated results showed an interesting trend for the deprotonation process, which would eventually control the isomerization of these alkenyl aromatics. Going from estragole to safrole, the pKa of H1a has reduced by several units. Thus, the deprotonation process of H1a would start competing with the allyl deprotonation H1 and hence the conversion would decrease on going down from estragole to safrole. The observed conversion in these cases supports the above rationalization for the isomerization process. Interestingly, the calculated pKa of H1a was found to be lower than the pKa of H1 for eugenol, which suggests that there should be minimal conversion in the isomerization process (Table 4) as there would be strong interference in the abstraction of allyl protons by the hydroxyl proton. Indeed, the least conversion (19%) for isomerization of eugenol observed over MgAl4 catalyst corresponded well with this theoretical prediction.
The experimentally observed results show that the isomerization leads to trans product as the major isomer in all cases. Table 5 gives the gas phase relative energies calculated at B3LYP/6-31+G* level of theory of the trans and cis products with respect to the corresponding reactant molecules. The calculated energies show that the trans products are energetically more favorable than the cis products which is in accordance to the experimental results. Recently, the study performed with platinum(II) complexes catalyzed isomerization of allylbenzenes also showed that the trans isomer is kinetically and thermodynamically favored over the cis-isomer.28 Solvent phase calculations performed with the PCM solvation model with DMF as the solvent (dielectric constant 38.2) also showed similar results (Table 5).
| Method | Trans- product | Cis- product |
|---|---|---|
| Allylbenzene |

|
|
| B3LYP/6-31+G* | −6.29 (−6.63) | −3.58 (−3.49) |
| Selectivity (%) | 88 | 12 |
| Estragole |
|
|
| B3LYP/6-31+G* | −6.47 (−6.80) | −3.39 (−3.36) |
| Selectivity (%) | 83 | 17 |
| Allylveratrole |
|

|
| B3LYP/6-31+G* | −6.23 (−6.61) | −3.37 (−3.41) |
| Selectivity (%) | 81 | 19 |
| Safrole |
|

|
| B3LYP/6-31+G* | −5.95 (−6.60) | −2.89 (−3.16) |
| Selectivity (%) | 87 | 13 |
| Eugenol |
|
 as as |
| B3LYP/6-31+G* | − 6.47 (−6.78) | −4.02 (−4.04) |
| Selectivity (%) | 81 | 19 |
Conclusion
Isomerization of alkenyl aromatics over series of MgAl and NiAl layered double hydroxides was successfully carried out under microwave irradiation. Under optimized conditions, MgAl4 showed 99% conversion of estragole to anethole with 86% trans selectivity. Significant variation in the conversion was noted under identical conditions for thermal heating. The trans form is more favorable than the cis form in all the cases. A good correlation was observed between the activities of the catalysts and the basicity derived using Hammett measurements. High-boiling-high-polar solvents like DMF, DMSO, and DMA gave high conversion (95 ± 3%) in shorter time (1 h), while high boiling-low polar, low boiling-high polar and low boiling-low polar solvents showed poor activity. The MgAl4 catalyst was recyclable for up to six cycles without significant activity loss. The isomerization of different alkenyl aromatics over MgAl4 showed substantial variation in the activity depending on the groups attached to the aromatic ring irrespective of the nature of heating. The relative pKa of the protons of the substituent groups attached to the alkenyl aromatics governs the conversion of the isomerization process. To summarise, reaction under microwave conditions offered both energy and environmental benefits by using reduced volume of solvent, shorter reaction time and at lesser reaction temperature.Acknowledgements
CMJ thanks CSIR, New Delhi for Senior Research Fellowship and AS thanks UGC, New Delhi for Senior Research Fellowship. The authors thank Analytical Science Discipline of this institute for analytical support.References
- (a) K. Baue, D. Garbe and H. Surberg, Flavors and Fragrances, in Ullmann's Encyclopedia of Industrial Chemistry, Electronic Release, 6th edn, 2002 Search PubMed; (b) International Flavors & Fragrances Inc., New Release, New York, N.Y., January 28, 2004.
- (a) A. Y. Leung, Encyclopedia of Common Natural Ingredients Used in Food, Drug and Cosmetics, John Wiley & Sons, New York, 1980 Search PubMed; (b) G. H. Eirew, U.S. Pat.Appl., US 2009035229, 2009 Search PubMed; (c) V. V. Kouznetsov and D. R. Merchan Arenas, Tetrahedron Lett., 2009, 50, 1546 CrossRef CAS; (d) A. J. Chalk, in Flavors and Fragances: A World Perspective, ed. B. M. Lawrence, B. D. Mookherjee and B. J. Willis, Elsevier Science, Amsterdam, 1988 Search PubMed.
- (a) L. Cerveny, A. Krejeikova, A. Marhoul and V. Ruzicka, React. Kinet. Catal. Lett., 1987, 33, 471 CrossRef CAS; (b) A. Loupy and L. Thach, Synth. Commun., 1993, 23, 2571 CrossRef CAS.
- N. Kameda and T. Yoneda, Nippon Kagaku Kaishi, 2000, 10, 677 CrossRef.
- (a) B. Lastra-Barreira, J. Francos, P. Crochet and V. Cadierno, Green Chem., 2011, 13, 307 RSC; (b) B. Lastra-Barreira and P. Crochet, Green Chem., 2010, 12, 1311 RSC.
- (a) H. Hattori, Chem. Rev., 1995, 95, 537 CrossRef CAS; (b) K. Tanabe and W. F. Holderich, Appl. Catal., A, 1999, 161, 399 CrossRef; (c) I. Al-Maskery, K. Girling, S. D. Jackson, L. Pugh and R. R. Spence, Top. Catal., 2010, 53, 1163 CrossRef CAS.
- (a) D. Kishore and S. Kannan, J. Mol. Catal. A: Chem., 2004, 223, 225 CrossRef CAS; (b) C. M. Jinesh, C. A. Antonyraj and S. Kannan, Catal. Today, 2009, 141, 176 CrossRef CAS; (c) C. M. Jinesh, V. Rives, D. Carriazo, C. A. Antonyraj and S. Kannan, Catal. Lett., 2010, 134, 337 CrossRef CAS.
- (a) S. Kannan, Catal. Surv. Asia, 2006, 10, 117 CrossRef CAS; (b) C. N. Perez, C. A. Perez, C. A. Henriques and J. L. F. Monteiro, Appl. Catal., A, 2004, 272, 229 CrossRef; (c) V. R. Choudhary, R. Jha and V. S. Narkhede, J. Mol. Catal. A: Chem., 2005, 239, 76 CrossRef CAS; (d) H. A. Prescott, Z. J. Li, E. Kemintz, A. Trunschke, J. Deutsch, H. Lieske and A. Auroux, J. Catal., 2005, 234, 119 CrossRef CAS; (e) C. A. Antonyraj and S. Kannan, Appl. Catal., A, 2008, 338, 121 CrossRef CAS; (f) C. M. Jinesh, C. A. Antonyraj and S. Kannan, Appl. Clay Sci., 2010, 48, 243 CrossRef CAS.
- X. Duan and D. G. Evans, Structure and Bonding-Layered Double Hydroxides, Springer, Berlin, 2006 Search PubMed.
- D. Kishore and S. Kannan, J. Mol. Catal. A: Chem., 2006, 244, 83 CrossRef CAS.
- (a) G. Bond, R. B. Moyes and D. A. Whan, Catal. Today, 1993, 17, 427 CrossRef CAS; (b) D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 1 RSC.
- (a) S. Vijaikumar and K. Pitchumani, Ind. J. Chem., 2010, 49B, 469 CAS; (b) U. R. Pillai, E. Sahle-Demessie and R. S. Varma, Tetrahedron Lett., 2002, 43, 2909 CrossRef CAS.
- L. N. Thach and C. R. Strauss, J. Chem., 2000, 38, 76 Search PubMed.
- Joint Committee on Powder Diffraction Standards, International Centre for Diffraction Data, Pennsylvania, USA, 1996, Set No. 46 Search PubMed.
- D. Kishore and S. Kannan, Green Chem., 2002, 4, 607 RSC.
- W. Xie and X. Huang, Catal. Lett., 2006, 107, 53 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Molecular Orbital Theory, Wiley, New York, 1988 Search PubMed.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210 CrossRef CAS; (d) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS; (e) M. Cossi and V. Barone, J. Chem. Phys., 1998, 109, 6246 CrossRef CAS.
- (a) G. Schüürmann, Chem. Phys. Lett., 1999, 302, 471 CrossRef; (b) M. W. Palascak and G. C. Shields, J. Phys. Chem. A, 2004, 108, 3692 CrossRef CAS; (c) G. I. Almerindo, D. W. Tondo and J. R. Pliego Jr, J. Phys. Chem. A, 2004, 108, 166 CrossRef CAS; (d) Z. K. Jia, D. M. Du, Z. Y. Zhou, A. G. Zhang and R. Y. Hou, Chem. Phys. Lett., 2007, 439, 374 CrossRef CAS; (e) C. Lim, D. Bashford and M. Karplus, J. Phys. Chem., 1991, 95, 5610 CrossRef CAS.
- D. M. Camaioni and C. A. Schwerdtfeger, J. Phys. Chem. A, 2005, 109, 10795 CrossRef CAS.
- I. A. Topol, G. J. Tawa, S. K. Burt and A. A. Rashin, J. Phys. Chem. A, 1997, 101, 10075 CrossRef CAS.
- K. Daasbjerg, Acta Chem. Scand., 1995, 49, 878 CrossRef CAS.
- (a) T. N. Brown and N. Mora-Diez, J. Phys. Chem. B, 2006, 110, 9270 CrossRef CAS; (b) A. Trummal, A. Rummel, E. Lippmaa, P. Burk and I. A. Koppel, J. Phys. Chem. A, 2009, 113, 6206 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) A. Brito, M. E. Borges, M. Garin and A. Hernandez, Energy Fuels, 2009, 23, 2953 CrossRef; (b) D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183 CrossRef CAS.
- S. K. Jana, P. Wu and T. Tatsumi, J. Catal., 2006, 240, 268 CrossRef CAS.
- A. Scarso, M. Colladon, P. Sgarbossa, C. Santo, R. A. Michelin and G. Strukul, Organometallics, 2010, 29, 1487 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR and TG-DTG of the MgAlx and NiAlx LDHs. See DOI: 10.1039/c2ra20179h |
| This journal is © The Royal Society of Chemistry 2012 |