Synthesis of molecular photoswitches based on azobenzene with an organosilane anchor†
Stephanie
Möller
,
Uwe
Pliquett
and
Christian
Hoffmann
*
Institute for Bioprocessing and Analytical Measurement Techniques, Rosenhof, 37308, Heilbad Heiligenstadt, Germany. E-mail: christian.hoffmann@iba-heiligenstadt.de; Fax: +49 3606 671200; Tel: +49 3606 671600
First published on 22nd March 2012
Abstract
The incorporation of photoactive molecules in thin layers enables photoinduced changes in wettability, e.g. azobenzenes, or micropatterning by deprotection of functional groups, e.g. applying nitroveratryl compounds. This paper describes a synthetic route for obtaining an azobenzene with a silane anchor. The chemical synthesis, including all intermediates, is characterized by NMR and IR. The photoisomerization of all products was investigated by UV/Vis spectroscopy. Ellipsometry and contact angle measurements give information about monolayers of the synthesized organosilanes.
Introduction
The application of light as a trigger for inducing changes in a molecular assembly is very attractive for release of chemical compounds,1,2 photopatterning3 and tuning wettability.4 Photoisomerization has been used as a molecular switch for many purposes. The simplest chemical compounds for this application are the derivates of azobenzene (C6H5N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH5C6). Irradiation in the UV wavelength range induces a conformational change from the thermodynamically more stable trans to the cis isomer. Owing to the reversibility of the reactions, irradiation with blue light switches the molecule back to the trans isomer.5 Another way of inducing the conformational change can be realized by electronic excitation. Henzl et al. performed the isomerization on an Au(111)-surface by positioning the tip of a scanning tunneling microscope and transferring the energy via inelastic tunneling electrons.6
NH5C6). Irradiation in the UV wavelength range induces a conformational change from the thermodynamically more stable trans to the cis isomer. Owing to the reversibility of the reactions, irradiation with blue light switches the molecule back to the trans isomer.5 Another way of inducing the conformational change can be realized by electronic excitation. Henzl et al. performed the isomerization on an Au(111)-surface by positioning the tip of a scanning tunneling microscope and transferring the energy via inelastic tunneling electrons.6
The required wavelength, i.e. energy, for the isomerization depends on the nature of the substituent R and the chemical environment.7,8 The activation energy EA for this process can be calculated by the Arrhenius equation. In addition to the substituent R, the character of the solvent influences the activation energy. Chemical architectures with this kind of photochromic system have been applied for changing the wettability of surfaces,9,10 conversion of light energy into mechanical work11,12 and pharmaceutical drug release in nanogels.13,14 Laloyaux et al. described the light driven motion of an oil droplet at the water/air interface. The light radiation changes the chemical potential of an azobenzene based surfactant on the water surface inducing the liquid motion of the oil droplet along the gradient of surface tension.15 Additional applications promise azobenzene derivatives as reversible, chiral switches.16
The versatile applications of photo switches in supramolecular systems in solution and on solid surfaces or in polymers was illustrated by Hecht et al.5 Ichimura et al. demonstrated the motion of ∼2 μl oil droplets along surfaces via photo induced local control of wettability.4
For the chemical modification of surfaces, thin films can be produced by a self-assembly process.17,18 Thiol terminated compounds adsorb to gold surfaces,19,20 whereas organosilanes are typically used for the modification of silicon oxide surfaces.21,22
For an azobenzene containing chemical modification of gold surfaces, Pace et al. synthesized 4′-(biphenyl-4-ylazo)biphenyl-4-thiol in seven steps from 4-nitrosobiphenyl9 and Shin et al. synthesized 11-(4-phenylazo-phenoxy)undecan-1-thiol from 4-hydroxyazobenzene in three steps.23 Yang et al. modified aminosilane functionalized SiO2 surfaces by reaction with azobenzene acid-chloride.10
The present paper describes the synthetic route of azobenzene derivatives containing a silane anchor group for reaction with SiO2 surfaces. These organosilanes facilitate the chemical modification of SiO2 with azobenzene containing layers in a one-step surface reaction. The activation energy for the photoisomerization of all synthesized azobenzene compounds was determined in solution by UV/Vis spectroscopy. Thin films of azobenzene based organosilanes were produced and subsequently characterized by ellipsometry and contact angle measurements.
Results and discussion
Three azobenzene based organosilanes for modification of SiO2-surfaces have been investigated with different terminal functionalities: phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (PAS); 4-(4′-pentyl)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (PPAS) and 4-(4′-pentyloxy)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (POPAS). PAS with the end phenyl ring of the azobenzene entity (Fig. 1) exposed at the surface, has been synthesized in two steps. Starting with 4-phenylazophenol (PAP, 4-hydroxyazobenzene) as the nucleophile, it reacts with 11-bromoundecene via bimolecular nucleophilic substitution (SN2) to 11-(4-phenylazo-phenoxy)-undecene (PAPU). The silane anchor was introduced by hydrosilylation at the terminal double bond. Additionally, other organosilanes, 4-(4′-pentyl)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (PPAS) and 4-(4′-pentyloxy)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (POPAS), were synthesized from either 4-pentylaniline or 4-pentyloxyaniline in three steps: azo coupling, SN2 reaction, and hydrosilylation. All products were characterized by 1H-NMR (Fig. 1), 13C-NMR, IR, UV/Vis, DC and GC-MS (refer to electronic supplementary information†) and the activation energy in different solvents was calculated (Table 1).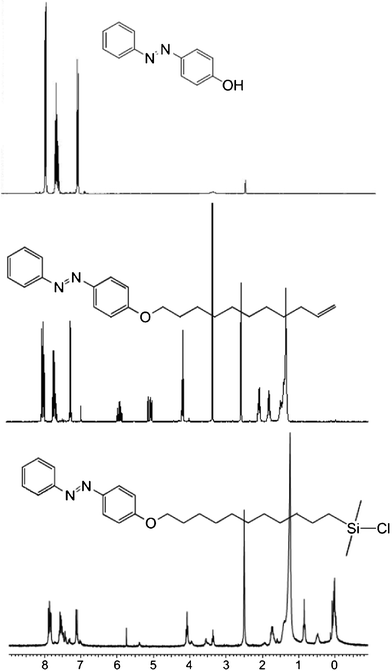 | ||
| Fig. 1 Synthetic route of an azobenzene compound with a silane anchor and characterization by 1H-NMR. Starting with 4-hydroxyazobenzene, an alkyl chain with a terminal double bond is introduced by a SN2 reaction and subsequently converted to a silane anchor group by hydrosilylation. | ||
| Molecule | E A(B)/kJ mol−1 | E A(I)/kJ mol−1 | k18(B)/10−4 s−1 | k18(I)/10−4 s−1 |
|---|---|---|---|---|
| PAP | — | 23 ± 2 | — | 0.3 ± 0.02 |
| PAPU | 109 ± 15 | 19 ± 3 | 0.4 ± 0.05 | 2.2 ± 0.02 |
| PAS | — | 32 ± 2 | — | 0.6 ± 0.02 |
| PPAP | 45 ± 3 | 47 ± 5 | 0.9 ± 0.09 | 0.8 ± 0.02 |
| PPAPU | 103 ± 2 | 55 ± 5 | 1.02 ± 0.1 | 0.5 ± 0.03 |
| PPAS | — | 28 ± 5 | — | 0.9 ± 0.09 |
| PPAS (back reaction) | — | 81 ± 5 | — | 0.4 ± 0.03 |
| POPAP | 82 ± 3 | 43 ± 5 | 0.08 ± 0.01 | 0.3 ± 0.03 |
| POPAPU | 24 ± 2 | 25 ± 5 | 0.39 ± 0.06 | 0.12 ± 0.01 |
| POPAPU (back reaction) | 66 ± 5 | 12 ± 5 | 0.09 ± 0.01 | 0.2 ± 0.05 |
| POPAS | — | 48 ± 5 | — | 0.9 ± 0.09 |
| POPAS (back reaction) | — | 64 ± 5 | — | 0.5 ± 0.03 |
The photo-isomerization speed of the synthesized compounds was investigated. Since it depends on multiple parameters, all environmental variables such as the concentration of the educts were kept constant within all sets, with exception of the parameter under test such as temperature and illumination. For comparison of the results between different sets of the experiments we always used the same wavelength λ = 380 nm and illumination intensity (2 mW cm−2).24 The molar extinction coefficients are different, so the solution concentrations were about 0.1 mM in the case of PPAP, PPAPU, PPAS and about 3 mM in the case of PAP, PAPU and PAS. To investigate the activation energy, EA, the concentration of the solutions for each molecule were kept constant at different temperatures.
Although it can be argued that the Arrhenius equation is not fully applicable, an apparent activation can be calculated from the linear dependence of the logarithm of the speed constant on the reciprocal of the temperature.25
The condition for the strict applicability of the Arrhenius equation is that the activation energy does not depend on temperature. However, the pre-exponential factor may depend on several factors like illumination density or concentration of the molecule under investigation.26
We have to distinguish between the reaction under illumination and without illumination. The cis–trans reaction has a negative Gibbs free energy and therefore runs spontaneously. In contrast, the reaction in the other direction is practically impossible without illumination.
To assess the activation energy of the spontaneous direction, the molecules were investigated between illumination for the cis-conformation and relaxation back to the trans-conformation at different temperatures. Under this condition neither light intensity nor concentration influences the reaction speed and a real activation energy can be found.
Switching under illumination is governed by the absorption of a photon. Since both the photon density (intensity of the light) and the absorption cross section of the molecules are basically independent of the temperature no temperature dependence of the reaction is expected. However, the experiment shows a clear dependence of the speed constant on the temperature, yielding an apparent activation energy that is not simply justified by the impact theory of chemical reactions.
This kind of activation energy, EA, was calculated using the Arrhenius equation for all steps. The photo isomerization reaction can be characterized by a first-order reaction. The Lambert–Beer law expresses the linear relationship between absorbance and concentration as described by Gauglitz et al.27,28 Therefore, the rate constants k were determined by recording UV/Vis spectra in solution during irradiation of the sample at 380 nm. The temperature for determining the rate constants was between 18 °C and 35 °C. While irradiating the sample at a wavelength of 380 nm the band at 380 nm assigned to an n→π* transition in the trans conformation decreases whereas the band at 440 nm assigned to an n→π* transition in the cis conformation increases (Fig. 2).
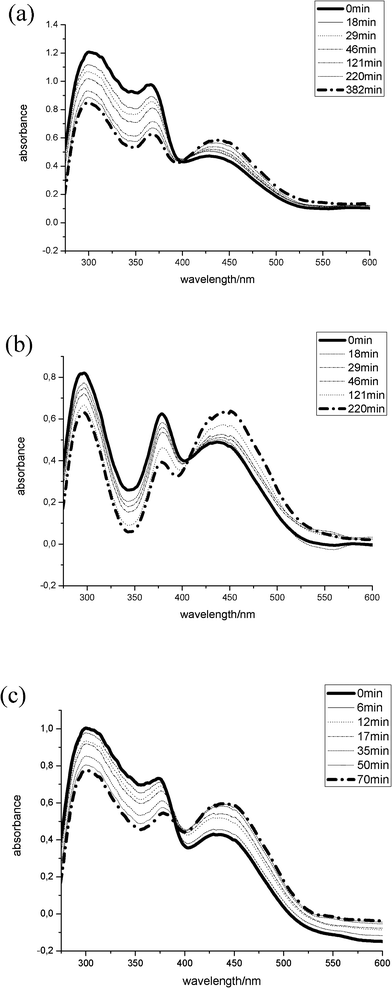 | ||
| Fig. 2 UV/Vis spectra for determination of kinetic rate k of photoisomerization during irradiation at λ = 380 nm of (a) 4-hydroxyazobenzene, c = 4.8 mM, (b) 11-(4-(phenylazo-phenoxy)-undecene, c = 2.5 mM, and (c) 4-phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)-benzene, c = 3.3 mM, in isooctane. | ||
The isomerization rate constant k and subsequently the apparent activation energy EA was determined at 380 nm. The main factor affecting the reaction speed under illumination is the probability of photonic absorption by a molecule. Especially in the trans to cis transition, once the photon is absorbed and the molecule is excited, a shorter time than that for spontaneous relaxation back to the non-excited state is needed to switch the molecule into a higher energy state.
The switching speed is mostly influenced by the viscosity of the solvent.29 A high viscosity and a long-tailed functional group at the azo moiety extends the time for the motion into the new position. At higher temperatures the viscosity decreases and the motion becomes faster. Finally, the experiments do not assess real switching speed but rather the probability that a photon is absorbed and yields the switch into the higher state.
In the other direction, from cis to trans, an absorbed photon has a higher probability for switching the molecule because of the higher Gibbs energy. In other words, the trans conformation is more likely because there are more possible configurations. However, as in case of the trans–cis reaction, if the excited molecule relaxes without switching, the molecule has to switch spontaneously or absorb another photon. This will clearly reduce the reaction speed.
We used sufficiently low concentrations, where the sample absorbs only a small fraction of the photons. Thus, the probability of absorbing a photon does not depend on the concentration. Different solvents have been applied for investigating the influence of polarity. 1,4-Butanediol was selected as a polar solvent and isooctane was selected as a nonpolar solvent. The resulting rate constants at 18 °C together with the calculated apparent activation energy of educts (PAP) and synthesized products PAPU and PAS are shown in Table 1. The apparent activation energy of PAPU in 1,4-butanediol is much higher than in isooctane, indicating that the isomerization is remarkably slower in polar solvents. No values were determined for PAP and PAS because they were insoluble in 1,4-butanediol. The hydrosilylation of PAS leads to a higher polarity and raises the activation energy. The rate constants reflect the same trend. These results are in agreement with calculations by Dubini-Paglia et al. for the activation energy of azobenzene compounds in dibutylphthalate.7 The reverse reaction from the trans- to cis-conformation could also be observed by recording UV/Vis spectra. However, the reaction velocity was too slow to obtain reasonable values for the apparent activation energy of PAS and PPAS.24
The reversibility of the POPAS isomerization can be followed by UV/Vis spectroscopy, indicating that different functional groups have an influence on the stability of the cis-conformation. Introduction of the pentoxy group causes a decrease in the activation energy of the re-isomerization and favors the back reaction, presumably due to the electron acceptor property of the ether group.7
The differences in the recorded rate constants k can also be explained by different mechanisms of photo isomerization. The possible mechanisms are shown in Fig. 3. Crecca et al.30 describes two pathways: the rotation occurs by out of plane torsion of the CNNC dihedral angle and the inversion undergoes in-plane bending of the NNC angle between the azo group and phenyl ring. Electron donating substituents increase the isomerization barrier along the inversion pathway. In a non polar solvent the barrier decreases after introducing an alkyl chain (PAP to PAPU) which means that in this case isomerization by the rotation pathway is more likely.30
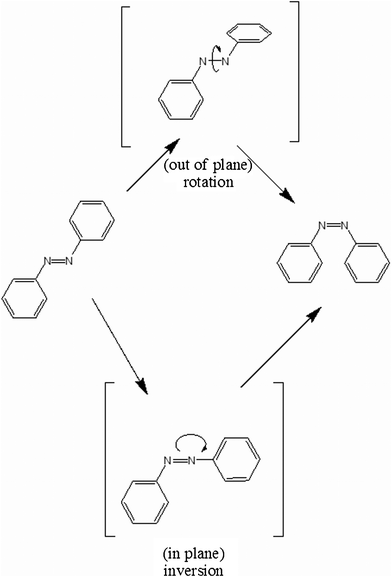 | ||
| Fig. 3 Schematic drawing of the two different mechanisms for photoisomerization of azobenzene derivates. It shows the rotation and inversion pathways of the azo group. | ||
Introducing a terminal pentyl group at the azobenzene entity leads to higher activation energies of the single products; probably owing to the large alkyl chains that have to be moved in the solvent while bending. The values for PAS, PPAS and POPAS obtained in isooctane are very similar. Since none of the compounds was soluble in 1,4-butanediol the activation energy could not be determined in this solvent. For higher significance of the experimental estimation of the activation energy we used especially 4-(4′-pentyloxy)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (POPAS) for assessment of the temperature dependent reaction speed from cis to trans under illumination but also as a spontaneous reaction without illumination. The difference in the apparent activation energy was found to be ΔE = 6 kJ mol−1. Measuring the reverse direction is impossible because the trans–cis conformational change does not proceed spontaneously.
In the case of azobenzene, the energy of the cis-conformation is 50 kJ mol−1 (12 kcal mol−1) higher than the trans-conformation. The activation energy in solution was 96 kJ mol−1 (23 kcal mol−1).31
The influence of irradiation on the rate constant k can be seen in Fig. 4. After 700 min and irradiation of the sample for 100 min at 380 nm the light was turned off resulting in a decreased rate constant k for the re-isomerization. Furthermore, the Arrhenius plots for that compound illustrate exemplarily the determination of the apparent activation energy.
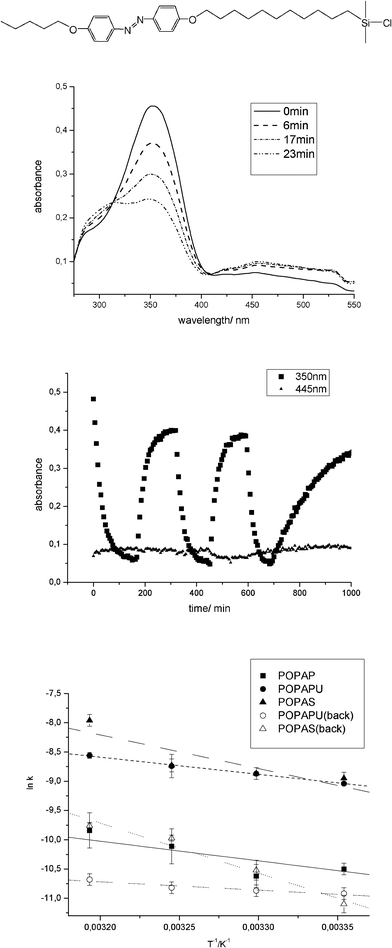 | ||
| Fig. 4 Reversibility of photoisomerization in solution (exemplarily at 40 °C). During irradiation at 380 nm in isooctane the absorbance at 350 nm decreases and during irradiation at 440 nm the absorbance increases at 350 nm. After 700 min the light was switched off and the rate constant k decreases. Plotting the logarithm of the rate constant based on the absorption spectra against the reciprocal temperature lead to the apparent activation energy determined by the slope. | ||
SiO2 surfaces have been coated with PAS, PPAS and POPAS by self-assembly and characterized by ellipsometry and contact angle measurements. The thickness of the PAS layer in the trans conformation was determined to be 0.9 nm ± 0.1 nm. This corresponds to 60% of the theoretical thickness (calculation based on bond lengths) taking into account that only 60% of an organosilane self-assembled monolayer,32,33 in case of PAS 2.6 nm, can be obtained (Fig. 5a). On the other hand, a dense monolayer of 2.9 nm ± 0.6 nm can be assembled from PPAS, as compared to the calculated thickness of 3.28 nm (Fig. 5b). This indicates that the introduction of the pentyl chain causes stronger van der Waals interactions. For POPAS an inhomogeneous layer 3.6 nm ± 2.1 nm thick (theoretical thickness 3.52 nm) was achieved (Fig. 5c). Therefore, the inserted oxygen atom may disturb the assembly process. The incomplete assembly compared to the theoretical layer thickness is based on the fact that the two methyl groups at the silane anchor cause steric effects. Furthermore, they avoid any cross-linking between the silane anchor groups.
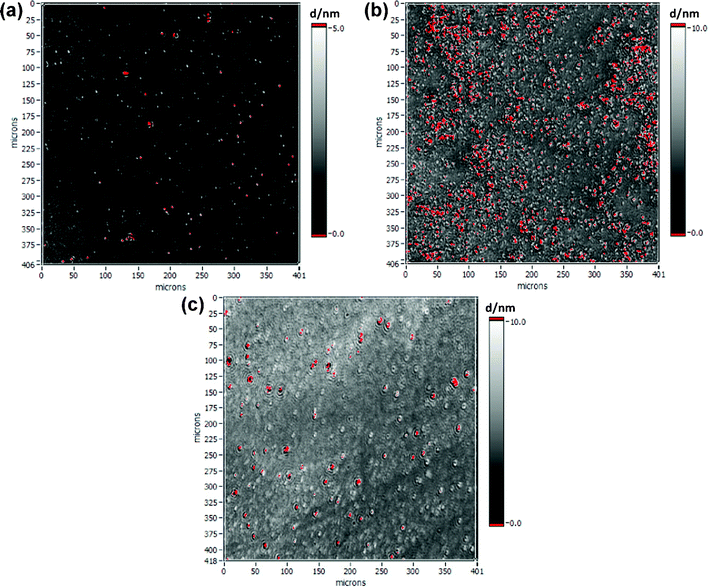 | ||
| Fig. 5 Imaging ellipsometry of surface modification on SiO2 (silicon wafer). Characterization after deposition of (a) 4-phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (PAS), (b) 4-(4′-pentyl)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (PPAS) and (c) 4-(4′-pentyloxy)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene (POPAS), at λ = 532 nm; n(Si) = 4.1653; k(Si) = 0.049; n(SiO2) = 1.4605; k(SiO2) = 0; d(SiO2) = 2.2 nm ± 0.7 nm (measured); n(organosilane) = 1.5; k(organosilane) = 0. | ||
For the motion of droplets on surfaces only a small contact angle hysteresis is tolerated since this application requires that the advancing contact angle θadv of the hydrophilic area must be smaller than the receding contact angle θrec of the hydrophobic region of the surface in the case of H2O as the liquid. The contact angles were measured with droplets of polar and non-polar solvents and the hysteresis was calculated. Tables 2–4 summarize the contact angles of various solvents on trans-azobenzene modified surfaces. The smallest hysteresis was found for diiodomethane on the PAS covered surface and increases with polarity where water yields the highest hysteresis. This trend is in good accordance with the contact measurements of Yang et al. on SiO2 surfaces modified with an azobenzene consisting layer.10 The higher the polarity of the solvent, the higher the contact angle and its hysteresis of the PAS covered surface. The smallest hysteresis of 20° has been obtained on a PAS covered surface applying a diiodomethane droplet.10 Irradiation causes a droplet to move if the change in contact angle due to azobenzene isomerization is higher than the hysteresis. Since the contact angle hysteresis increases with the surface roughness the homogeneity of the layer is of crucial importance. There are two ways of meeting the requirements for droplet motion: producing surfaces with low roughness and thus small contact angle hysteresis, or enhancing the contact angle change due to the isomerization process. The latter can be achieved by the introduction of terminal hydrophobic groups, e.g. fluoride. The observed difference between θadv and θrec of non-polar solvents in the literature ranges from 4° to 15°. Yang et al. observed the motion of benzonitrile droplets as a non polar solvent and a contact angle hysteresis of 6°.
| Liquid | θ adv/° | θ rec/° | Hysteresis/° |
|---|---|---|---|
| Water | 86 ± 10 | 26 ± 3 | 60 ± 10 |
| Droplet of water covered with tetradecane | 104 ± 5 | 36 ± 2 | 68 ± 3 |
| Ethyleneglycol | 62 ± 2 | 21 ± 2 | 41 ± 2 |
| Diiodomethane | 60 ± 4 | 40 ± 3 | 20 ± 4 |
| Formamide | 72 ± 1 | 18 ± 1 | 54 ± 1 |
| Liquid | θ adv/° | θ rec/° | Hysteresis/° |
|---|---|---|---|
| Water | 101 ± 1 | 70 ± 10 | 31 ± 10 |
| Droplet of water covered with tetradecane | 140 ± 5 | 84 ± 5 | 56 ± 5 |
| Ethyleneglycol | 91 ± 9 | 55 ± 10 | 36 ± 10 |
| Diiodomethane | 83 ± 2 | 51 ± 7 | 32 ± 5 |
| Formamide | 94 ± 3 | 50 ± 3 | 44 ± 3 |
| Liquid | θ adv/° | θ rec/° | Hysteresis/° |
|---|---|---|---|
| Water | 87 ± 3 | 24 ± 1 | 63 ± 2 |
| Droplet of water covered with tetradecane | 134 ± 5 | 78 ± 20 | 56 ± 20 |
| Ethyleneglycol | 78 ± 5 | 17 ± 5 | 61 ± 5 |
| Diiodomethane | < 5 | < 5 | — |
| Formamide | 78 ± 3 | 24 ± 2 | 54 ± 3 |
To monitor the dependence of the contact angle θ of a droplet on the irradiation time, a PAS modified glass substrate was illuminated at λ = 380 nm from the back side and the static contact angle determined. The water droplet was covered with tetradecane to avoid evaporation effects. When the surface was irradiated the contact angle immediately decreased by 5° ± 1° and after 30 min irradiation the contact angle decreased by 7° ± 1°. The contact diameter of the droplet increased by 12%. The small effect may be due to the high density of the layer since the available space is not sufficient and inhibits isomerization. Therefore, the cis/trans-isomerization in a molecular layer works better at a water/air interface than on solid surfaces since the molecules can spread at the interface leading to more space for bending. Laloyoux and Jonas made an oil droplet move along an air/water interface by spreading an azobenzene based surfactant on the water surface.15 The photoinduced isomerization causes a surface tension gradient. The resulting convection at the droplet surface leads to its motion from areas with lower surface tension to areas with high surface tension.
A mixed layer consisting of azobenzene-containing organosilane compounds and short aliphatic organosilanes as a spacer in between may improve the isomerization on the surface. Additionally the application of platforms, consisting of an anchor group based on so-called triacatriangulinium ions (TATA+) with ethinyl or phenyl groups for further reaction with azobenzene derivatives may prove beneficial.34,35
Experimental
Determination of isomerization rate constants
A 1.2 ml quartz glass cuvette with an optical path length of 1 cm was filled with 0.25 cm3 sample solution. The cuvette was arranged in a temperature controlled box with four optical windows at 90° distance for two perpendicular light paths. A deuterium lamp served as light source and illuminates the sample with an optical fiber for recording the spectra. On the opposite side was a spectrometer (Ocean Optics QE65000) connected with another optical fiber. The light source for the photoisomerization consisted of a 100 W Xenon lamp, collimation optics and a monochromator that was arranged at 90° to the spectrometer beam and provided the required wavelength. The slit was adjusted to meet a bandwidth of 20 nm. The irradiation power of 2 mW cm−2 was measured using an optical powermeter. Spectra from 280 nm to 600 nm were taken every 5 min after calibration of the optical set-up by recording a baseline.The concentrations were about 0.1 mM in the case of PPAP, PPAPU, PPAS, POPAP, POPAPU, and POPAS and were about 3 mM in the case of PAP, PAPU and PAS.
Measurement of the contact angle
The illumination optics of an inverted fluorescence microscope (Olympus IMT2) was equipped with the corresponding filters for the photoisomerization of the chromophore (380 nm, 430 nm). A droplet of water was placed accurately in the illumination path in a quartz glass cuvette and covered with tetradecane. For contact angle measurements, a high power LED (LUXEON) illuminated the droplet and the shadow was recorded via a telescope objective by a video camera. The wavelength for the photoisomerization was set by the filter cartridge. The contact angle was determined on both sides of the droplet and the contact diameter was measured.Synthesis of 11-(4-phenylazo-phenoxy)-undecene
4 mmol 4-hydroxyazobenzene was dissolved in 10 ml of dimethyl formamide (DMF), 5.8 mmol potassium carbonate. 1 mmol of the catalyst dicyclohexano-18-crown-6 was added, and the solution was heated to 100 °C. A solution of 4 mmol 11-bromoundecene in 5 ml of DMF was added dropwise. The reaction took place overnight at 100 °C. The solvent was removed under reduced pressure. The residue was dissolved in 20 ml of dichloromethane and washed with water. The solvent was removed and the product was purified by chromatography using a silica gel column (eluent diethyl ether/n-hexane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9).
:9).
Orange crystals, yield 97%, GC purification grade 97%, about 8% dicyclohexano-18-crown-6 detected in 1H-NMR that could not be removed, retention time (GC, polyphenylmethylsiloxane-column 30 m, DB-5 MS, J&W Scientific) t = 35 min. Since the azobenzene compounds decompose under elevated temperature further purification was not feasible.
Mp 67 °C, Rf (diethyl ether/hexane 1:9) = 0.6. 1H NMR (300 MHz, DMSO) δ [ppm]: 1.1–1.5 (m, 12 H, CH2CHCH2(CH2)6CH2), 1.7 (m, 2 H, CH2CH2CH2O), 2.0 (dt, 2H, CH2CHCH2CH2), 2.5 (DMSO), 3.3 (dicyclohexano-18-crown-6), 4.1 (t, 2H, CH2CH2O), 4.9–5.1 (m, 2H, CH2CHCH2), 5.8 (m, 1H, CH2CHCH2), 7.1 (d, 2H, Ph-H), 7.5 (m, 3H, Ph-H), 7.8 (2 × d, 4H, Ph–H). 13C NMR (126 MHz, DMSO) δ [ppm] 25.35 (1 C, CH2CH(CH2)4CH2), 28.18–28.94 six signals (6 C, CH2), 33.05 (1 C, CH2CHCH2), 39.5–39.83 (DMSO), 67.88 (1 C, CphenylOCH2CH2), 114.37 (1 C, CH2CHCH2), 114.83 (2 C, CphenylH), 121.98 (2 C, CphenylH), 124.33 (2 C, CphenylH), 129.11 (2 C, CphenylH), 130.50 (1 C, terminal CphenylH), 138.57 (1 C, CH2CHCH2), 145.82 (1 C, CphenylNN), 151.80 (1 C, CphenylNN), 161.24 (1 C, CphenylOCH2). IR (KBr) [cm−1]: 800–840 m, (1,4-disubstituted aromatic compound); 1250 s, ν(C–O) alkylaryl ether; 1460 m, δ(CH2), 1520–1680 m, ν(CC), aromatic compound; 2851 s, νs(CH2); 2926 s, νas(CH2); 3069 w, ν(C–H), aromatic compound. MS (70 eV): m/z (%): 350 (8) [M–H]+, 121 (20) [C9H12]+, 107 (100) [C6H7N2]+, 77 (56) [C6H6]+, 41 (80) [C3H5]+, 39 (16) [C3H3]+. UV/Vis (isooctane): λmax (ε) = 297 nm (328), 379 nm (248), 450 nm (256).
Synthesis of 4-phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene
Under argon atmosphere 0.1 mmol 11-(4-phenylazo-phenoxy)-undecene was dissolved in 10 ml of distilled dichloromethane and a spatula tip of platinum catalyst added. After addition of 0.5 mmol dimethylchlorosilane the solution was stirred for about 18 h at RT under reflux. The solvent and excess dimethylchlorosilane were removed under reduced pressure. The residue, consisting of the product and traces of catalyst, was dried.Yellow crystals, quantitative yield including catalyst (may be separated by reaction at the surface). Mp 56 °C (decomposition). 1H NMR (300 MHz, DMSO); δ [ppm]: 0 (silyl residues), 0.5 (s, 6 H, ClSi(CH3)2CH2), 0.8 (t, 2 H, ClSi(CH3)2CH2CH2), 1.1–1.5 (m, 16 H, ClSi(CH3)2CH2(CH2)8CH2CH2O), 1.7 (m, 2 H, CH2CH2CH2O), 2.5 (DMSO), 3.3 (dicyclohexano-18-crown-6), 4.1 (t, 2 H, CH2CH2O), 7.1 (d, 2 H, Ph–H), 7.5 (m, 3 H, Ph–H), 7.8 (2 × d, 4 H, Ph–H). 13C NMR (126 MHz, CDCl3) δ [ppm] 0.33 (silyl residues), 0.51 (2 C, SiCH3), 18.51 (1 C, SiCH2), 22.76–33.51 eight signals (9 C, CH2), 68.41 (1 C, CphenylOCH2CH2), 77.00 (CHCl3), 114.65 (2 C, CphenylH), 122.46 (2 C, CphenylH), 124.74 (2 C ,CphenylH), 128.93 (2 C, CphenylH), 130.21 (1 C, terminal CphenylH), 146.68 (1 C, CphenylNN), 152.59 (1 C, CphenylNN), 161.66 (1 C, CphenylOCH2). IR (KBr) [cm−1]: 800–840 m, (1,4-disubstituted aromatic compound); 1100 s, ν(Si–O), due to hydrolysis of Si–Cl during KBr preparation; 1250 s, ν(C–O) alkylaryl ether; 1460 m, δ(CH2), 1520–1680 m, ν(CC), aromatic compound; 2851 s, νs(CH2); 2926 s, νas(CH2); 2963 w, ν(CH3); 3069 w, ν(C–H), aromatic compound. UV/Vis (isooctane): λmax (ε)= 300 nm (303), 373 nm (221), 442 nm (182). GC-analysis was not possible since the silane anchor group adsorbs irreversibly to the stationary phase.
Synthesis of 4-(4′-pentyl)phenylazophenol
22 mmol 4-pentylaniline was dissolved in 9 ml of HClconc in 12 ml of water. While stirring and cooling, 69 mmol sodium nitrite was added dropwise holding the temperature constant at 0 °C. 22 mmol phenol and 3 mmol sodium carbonate was dissolved in 12 ml of ultrapure water. The prepared solution of diazonium salt was added dropwise and subsequently stirred for 30 min. The solution was stored overnight in the refrigerator at 4 °C for separation. Then the product was dissolved in 100 ml of diethyl ether and the aqueous phase was isolated by a separation funnel. The aqueous phase was extracted another three times with 50 ml of diethyl ether. The solvent was removed and the product purified by chromatography using silica gel (eluent diethyl ether/n-hexane 1:1).
Orange crystals, yield 35%, retention time (GC, polyphenylmethylsiloxane column 30 m, DB-5 MS, J&W Scientific) t = 24 min. Mp 77 °C, Rf (diethyl ether/n-hexane 1:1) = 0.76. 1H NMR (300 MHz, CDCl3); δ [ppm]: 0.89 (t, 3 H, CH3CH2CH2), 1.32 (m, 4 H, CH3CH2CH2), 1.64 (m, 2 H, CphenylCH2CH2), 2.65 (t, 2 H, CphenylCH2CH2), 6.87 (d, 2 H, Ph–H), 7.30 (d, 2 H, Ph–H), 7.81 (2 × d, 4 H, Ph–H). 13C-NMR (CDCl3, 126 MHz) δ [ppm] 14.40 (1 C, CH3CH2), 22.90 (1 C, CH3CH2CH2), 31.35 (1 C, CH3CH2CH2), 31.83 (1 C, CphenylCH2CH2), 36.19 (1 C, CphenylCH2CH2), 77.42 (CHCl3), 116.29 (2 C, CphenylH), 122.90 (2 C, CphenylH), 125.19 (2 C, CphenylH), 129.50 (2 C,CphenylH), 146.45 1 C, CphenylCH2), 147.33 (1 C, CphenylNN), 151.22 (1 C, CphenylNN), 158.83 (1 C, CphenylOH). IR (KBr) [cm−1]: 839 m, (1,4-disubstituted aromatic compound); 1250 s, ν(C–O) alkylaryl ether; 1389–1498 m, ν(CC), aromatic compound; 2853 s, νs(CH2); 2925 s, νas(CH2); 2956 s, νs(CH3); 3069 w, ν(C–H) aromatic compound, 3250 w, νs (O–H). MS (70 eV): m/z (%): 268 (35) [M–H]+, 147 (50) [C11H15N2]+, 121 (75) [C9H12]+, 93 (100) [C8H10]+, 65 (40) [C5H5]+, 39 (30) [C3H3]+. UV/Vis (isooctane): λmax (ε) = 290 nm (1784), 389 nm (2159), 430 nm (1289).
Synthesis of 4-(4′-pentyl)phenylazo-phenoxy)-undecene
4 mmol 4-(4′-pentyl)phenylazophenol was dissolved in 10 ml of dimethylformamide (DMF), 5.8 mmol potassium carbonate. 1 mmol of the catalyst dicyclohexano-18-crown-6 was added, and the solution was heated to 100 °C. 4 mmol 11-bromo undecene was dissolved in 5 ml of DMF and added dropwise into this solution. The reaction took place overnight at 100 °C. The solvent was removed under vacuum. The residue was dissolved in 20 ml of dichloromethane and washed with water. After removing the solvent, the product was purified by silica gel chromatography (eluent diethyl ether/n-hexane 1:9).
Orange crystals, yield 50%, GC-purification 93%, about 8% dicyclohexano-18-crown-6 that could not be removed was detected by 1H-NMR, retention time (GC, polyphenylmethylsiloxane column 30 m, DB-5 MS, J&W Scientific) t = 41 min. Since the azobenzene compounds decompose at high temperatures further purification was not feasible.
Mp 78 °C, Rf (diethyl ether/n-hexane 1:9) = 0.66. 1H-NMR (CDCl3, 300 MHz) δ [ppm] 0.88 (t, 3 H, CH3CH2), 1.32–1.46 (m, 12 H, CH2CHCH2(CH2)6CH2 and 4 H, CH3CH2CH2CH2), 1.64 (t, 2 H, CphenylCH2CH2), 1.7 (m, 2 H, CH2CH2CH2O), 2.01 (dt, 2 H, CH2CHCH2CH2), 2.65 (t, 2 H, CphenylCH2CH2), 4.01 (t, 2 H, CH2CH2O), 4.8–5.0 (m, 2 H, CH2CHCH2), 5.81 (m, 1 H, CH2CHCH2), 6.99 (d, 2 H, Ph–H), 7.24 (CHCl3), 7.29 (d, 2 H, Ph–H), 7.76 (d, 2 H, Ph–H), 7.85 (d, 2 H, Ph–H). 13C-NMR (CDCl3, 126 MHz) δ [ppm] 13.98 (1 C, CH3CH2), 22.48 (1 C, CH3CH2CH2), 25.97 (1 C, CH2CH(CH2)4CH2) 28.88–29.46 six signals (6 C, CH2), 30.96 (1 C, CH3CH2CH2), 31.43 (1 C, CphenylCH2CH2), 33.76 (1 C, CH2CHCH2), 35.78 (1 C, CphenylCH2CH2), 68.30 (1 C, CphenylOCH2CH2), 77.00 (CHCl3), 114.63 (1 C, CH2CHCH2), 114.56 (2 C, CphenylH), 122.47 (2 C, CphenylH), 124.51 (2 C, CphenylH), 128.98 (2 C, CphenylH), 139.16 (1 C, CH2CHCH2), 145.72 1 C, CphenylCH2), 146.90 (1 C, CphenylNN), 151.01 (1 C, CphenylNN), 161.43 (1 C, CphenylOCH2). IR (KBr) [cm−1]: 800–840 m, (1,4-disubstituted aromatic compound); 1021 1103 s, ν(C–O–C) sometimes split, alkylaryl ether; 1250 s, ν(C–O) alkylaryl ether; 1460 m, δ(CH2), 1520–1680 m, ν(CC), aromatic compound; 2851 s, νs(CH2); 2926 s, νas(CH2); 2963 s, ν(CH3); 3069 s, ν(C–H), aromatic compound. MS (70 eV): m/z (%):420 (8) [M–H]+, 147 (50) [C11H15N2]+, 121 (75) [C9H12]+, 93 (100) [C8H10]+, 65 (40) [C5H5]+, 39 (29) [C3H3]+. UV/Vis (isooctane): λmax (nm) = 297 nm (1006), 386 nm (940), 440 nm (242).
Synthesis of 4-(4′-pentyl)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene:
Under argon atmosphere 0.1 mmol 11-(4-(4′-pentyl)phenylazo-phenoxy)-undecene was dissolved in 10 ml of distilled dichloromethane and a spatula tip of platinum catalyst was added. After the addition of 0.5 mmol dimethylchlorosilane, the solution was stirred for about 18 h at RT under reflux. The solvent and excess dimethylchlorosilane were removed. The residue was dried under reduced pressure.Black oil, yield 58%, including catalyst and silyl residues. 1H NMR (300 MHz, CDCl3); δ [ppm]: 0.05 (silyl residues), 0.50 (s, 6 H, ClSi(CH3)2CH2), 0.87 (t, 2 H, Si(CH3)2CH2CH2) and (t, 3 H, CH3CH2), 1.26 (m, 16 H, ClSi(CH3)2CH2(CH2)8CH2CH2O and 4 H, CH3CH2CH2CH2), 1.63 (t, 2 H, CphenylCH2CH2), 1.79 (m, 2 H, CH2CH2CH2O), 4.01 (t, 2 H, CH2CH2O), 6.95 (d, 2 H, Ph–H), 7.26 (CHCl3), 7.29 (d, 2 H, Ph–H), 7.76 (d, 2 H, Ph–H), 7.86 (d, 2 H, Ph–H). 13C-NMR (CDCl3, 126 MHz) δ [ppm] 0,21 (silyl residues), 0.71–1.01 (2 C, SiCH3), 14.01 (1 C, CH3CH2), 18.27 (1 C, SiCH2), 22.52 (1 C, CH3CH2CH2), 23.23–31.45 eleven signals (11 C, CH2), 35.82 (1 C, CphenylCH2CH2), 68.37 (1 C, CphenylOCH2CH2), 76.75–77.25 (CHCl3), 114.68 (2 C, CphenylH), 122.50 (2 C, CphenylH), 124.60 (2 C, CphenylH), 129.03 (2 C, CphenylH), 145.79 1 C, CphenylCH2), 146.88 (1 C, CphenylNN), 150.97 (1 C, CphenylNN), 161.53 (1 C, CphenylOCH2). IR (KBr) [cm−1]: 802 m, (1,4-disubstituted aromatic compound); 1096 s, ν(Si–O, due to hydrolysis of Si–Cl during KBr preparation); 1024 s, ν(C–O–C); 1261s, ν(C–O) alkylaryl ether; 1466 m, δ(CH2), 1520–1608 m, ν(CC), aromatic compound; 2855 s, νs(CH2); 2925 s, νas(CH2); 2963 s, ν(CH3); 3069 w, ν(C–H), aromatic compound. UV/Vis (isooctane): λmax (ε) = 344 nm (733), 440 nm (200).
Synthesis of 4-(4′-pentyloxy)phenylazophenol
18 mmol 4-pentyloxyaniline was dissolved in 9 ml of HClconc in 12 ml of water. While stirring and cooling, 18 mmol sodium nitrite was added dropwise holding the temperature at about 0 °C during the whole time. 18 mmol phenol and 3 mmol sodium carbonate was dissolved in 12 ml of ultrapure water and the prepared solution of diazonium salt was added dropwise and subsequently stirred for 30 min. The solution was stored overnight in the refrigerator at 4 °C for separation. Then the product was dissolved in 100 ml of diethyl ether and the aqueous phase isolated by a separation funnel. The aqueous phase was extracted another three times with 50 ml of diethyl ether. The solvent was removed and the product purified by chromatography using silica gel (eluent diethyl ether/n-hexane 1:1).
Yellow crystals, yield 27%, retention time (GC, polyphenylmethylsiloxane column 30 m, DB-5 MS, J&W Scientific) t = 73 min. Mp 88 °C, Rf (diethyl ether/n-hexane/chloroform 3:4:1) = 0.86. 1H NMR (300 MHz, CDCl3); δ [ppm]: 0.88 (t, 3 H, CH3CH2CH2), 1.35 (m, 4 H, CH3CH2CH2), 1.8 (m, 2 H, CphenylCH2CH2), 6.9 (d, 4 H, Ph–H), 7.81 (2 × d, 4 H, Ph–H). 13C-NMR (CDCl3, 126 MHz) δ [ppm] 14.10 (1 C, CH3CH2), 22.52 (1 C, CH3CH2CH2), 77.42 (CHCl3), 115.68 (4 C, CphenylH), 124.45 (4 C, CphenylH), 146.71 (2 C, CphenylCH2), 157.51 (4 C, CphenylNN), 161.15 (1 C, CphenylOH). IR (KBr) [cm−1]: 839 m, (1,4-disubstituted aromatic compound); 1238 s, ν(C–O) alkylaryl ether; 1389–1498 m, ν(CC), aromatic compound; 2862 s, νs(CH2); 2938 s, νas(CH2); 2957 s, νs(CH3); 3069 w, ν(C–H), aromatic compound; 3250 w, νs(O–H). MS (70 eV): m/z (%): 282 (35) [M–H]+, 242 (50) [C15H16N2O]+, 121 (75) [C9H12]+, 93 (100) [C8H10]+, 65 (40) [C5H5]+, 43 (30) [C3H7]+. UV/Vis (isooctane): λmax (ε) = 352 nm (3154), 361 nm (2846), 424 nm (185).
Synthesis of 4-(4′-pentyloxy)phenylazo-phenoxy)-undecene
0.53 mmol 4-(4′-pentyloxy)phenylazophenol was dissolved in 5 ml of dimethylformamide (DMF), and 0.7 mmol potassium carbonate and 0.1 mmol catalyst dicyclohexano-18-crown-6 were added. The solution was heated to 100 °C. 0.53 mmol 11-bromo undecene was dissolved in 5 ml of DMF and dropped into this solution. The reaction took place overnight at 100 °C. The solvent was removed under reduced pressure and the residue was dissolved in 20 ml of dichloromethane and washed with water. After removing the solvent, the product was purified by silica gel chromatography (eluent diethyl ether/n-hexane 1:9).
Orange crystals, yield 93%, GC-purification 100%, about 8% dicyclohexano-18-crown-6 that could not be removed was detected by 1H-NMR, retention time (GC, polyphenylmethylsiloxane column 30 m, DB-5 MS, J&W Scientific) t = 99 min. Since the azobenzene compounds decompose at high temperatures further purification was not feasible.
Mp 82 °C, Rf (diethyl ether/n-hexane 1:9) = 0.32. 1H-NMR (CDCl3, 300 MHz) δ [ppm] 0.92 (t, 3 H, CH3CH2), 1.33–1.6 (m, 12 H, CH2CHCH2(CH2)6CH2 and 4 H, CH3CH2CH2CH2), 1.7 (m, 2 H, CH2CH2CH2O), 2.0 (dt, 2 H, CH2CHCH2CH2), 2.6 (t, 2 H, CphenylCH2CH2), 4.0 (t, 2 H, CH2CH2O), 5.0 (m, 2 H, CH2CHCH2), 5.8 (m, 1 H, CH2CHCH2), 6.9 (d, 4 H, Ph–H), 7.24 (CHCl3), 7.8 (d, 4 H, Ph–H). 13C-NMR (CDCl3, 126 MHz) δ [ppm] 14.00 (1 C, CH3CH2), 22.44 (1 C, CH3CH2CH2), 26.00 (1 C, CH2CH(CH2)4CH2), 28.91–29.20 six signals (6 C, CH2), 33.79 (1 C, CH3CH2CH2), 33.79 (1 C, CH2CHCH2), 68.29 (1 C, CphenylOCH2CH2), 77.00 (CHCl3), 114.12 (1 C, CH2CHCH2), 114.64 (2 C, CphenylH), 124.27 (4 C, CphenylH), 139.18 (1 C, CH2CHCH2), 146.93 (2 C, CphenylNN), 161.15 (2 C, CphenylOCH2). IR (KBr) [cm−1]: 841 m, (1,4-disubstituted aromatic compound); 1046 s, ν(C–O–C) sometimes split, alkylaryl ether; 1246 s, ν(C–O) alkylaryl ether; 1466 m, δ(CH2), 1520–1642 m, ν(CC), aromatic compound; 2852 s, νs(CH2); 2924 s, νas(CH2); 2963 s, ν(CH3); 3069 s, ν(C–H), aromatic compound. MS (70 eV): m/z (%): 436 (20) [M–H]+, 258 (5) [C18H28N]+, 163 (47) [C11H14O]+, 107 (76) [C7H7O]+, 69 (35) [C5H5O]+, 55 (82) [C3H3O]+, 43 (100) [C3H7]+. UV/Vis (isooctane): λmax (nm) = 356 nm (13333), 436 nm (86).
Synthesis of 4-(4′-pentyloxy)phenylazo-(4′-(11-dimethylchlorosilanyl)-undecyloxy)benzene
Under argon atmosphere 0.62 mmol 11-(4-(4′-pentyloxy)phenylazo-phenoxy)-undecene was dissolved in 10 ml of distilled dichloromethane and a spatula tip of platinum catalyst was added. After the addition of 3 mmol dimethylchlorosilane, the solution was stirred for about 18 h at RT under reflux. The solvent and excess dimethylchlorosilane were removed. The residue was dried under reduced pressure.Black crystals, yield 100%, including catalyst and silyl residues. 1H NMR (300 MHz, CDCl3); δ [ppm]: 0.05 (silyl residues), 0.50 (s, 6 H, ClSi(CH3)2CH2), 0.95 (t, 2 H, Si(CH3)2CH2CH2) and (t, 3 H, CH3CH2), 1.29 (m, 16 H, ClSi(CH3)2CH2(CH2)8CH2CH2O and 4 H, CH3CH2CH2CH2), 1.82 (m, 4 H, CH2CH2CH2O), 4.04 (t, 4 H, CH2CH2O), 6.98 (d, 4 H, Ph–H), 7.26 (CHCl3), 7.93 (d, 4 H, Ph–H). 13C-NMR (CDCl3, 126 MHz) δ [ppm] 0.20–0.39 (silyl residues), 0.73–1.66 (2 C, SiCH3), 14.00 (1 C, CH3CH2), 18.42 (1 C, SiCH2), 22.44 (1 C, CH3CH2CH2), 23.29–33.43 eleven signals (11 C, CH2), 68.39 (1 C, CphenylOCH2CH2), 76.75–77.25 (CHCl3), 114.78 (4 C, CphenylH), 124.65 (4 C, CphenylH), 146.17 (2 C, CphenylNN), 161.55 (2 C, CphenylOCH2). IR (KBr) [cm−1]: 802 m, (1,4-disubstituted aromatic compound); 1095 s, ν(Si–O), due to hydrolysis of Si–Cl during KBr preparation; 1261 s, ν(C–O) alkylaryl ether; 1466 m, δ(CH2), 1511–1729 m, ν(CC), aromatic compound; 2854 s, νs(CH2); 2925 s, νas(CH2); 2963 s, ν(CH3); 3069 w, ν(C–H), aromatic compound. UV/Vis (isooctane): λmax (nm) = 356 nm (5850), 443 nm (100).
Surface modification of silicon wafer
After cleaning the silicon wafers with alkaline cuvette cleaner solution, they were activated with 25% NH3/30% H2O2 solution (3:1) heated to 75 °C for 30 min. After activation, the wafer was rinsed with ultrapure water and dried with argon. The activated and dried wafer was dipped in an organosilane solution for the self-assembly process (5 ml of absolute toluene, 20 μl triethyl amine and 0.04 mmol PAS or PPAS or POPAS) and reacted for 18 h under argon atmosphere. Then the wafers were removed from solution separately, rinsed with toluene, chloroform, ultrapure water and dried with an argon stream.
Conclusions
The presented synthetic route of photo switchable organosilanes shows a general way to synthesize chemical compounds that consist of an anchor group for SiO2 surfaces, an azobenzene entity for photoisomerization and a terminal functional group. Self assembly enables the production of photoactive surfaces as a one-step process. This might be advantageous compared to multi-step synthesis at the surface where the yield of the coating is generally smaller or the first layer partly desorbs while the second is added.Acknowledgements
The authors thank Reinhard Machinek from the Georg-August-University Göttingen for recording the NMR spectra, Dieter Frense (iba) for recording the GC-MS spectra, Nadja Ehrhardt (iba) for laboratory assistance, Brian P. Cahill (iba) for valuable discussions and the German BMBF for funding the project OSOKA (FKZ: 16SV3537).References
- K. Schmitt, J. Rist and C. Hoffmann, Anal. Bioanal. Chem., 2011, 401, 777–782 CrossRef CAS.
- X. Wang, S. Werner, T. Weiβ, K. Liefeith and C. Hoffmann, RSC Adv., 2012, 2, 156–160 RSC.
- J. M. Alonso, A. Reichel, J. Piehler and A. del Campo, Langmuir, 2008, 24, 448–457 CrossRef CAS.
- K. Ichimura, S.-K. Oh and M. Nakagawa, Science, 2000, 288, 1624–1626 CrossRef CAS.
- M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS.
- J. Henzl, M. Mehlhorn, H. Gawronski, K. H. Rieder and K. Morgenstern, Angew. Chem., Int. Ed., 2006, 45, 603–606 CrossRef CAS.
- E. Dubini-Paglia, P. L. Beltrame, B. Marcandalli, P. Carniti, A. Seves and L. Vicini, J. Appl. Polym. Sci., 1986, 31, 1251–1260 CrossRef CAS.
- E. Dubini-Paglia, P. L. Beltrame, B. Marcandalli, A. Seves and L. Vicini, J. Appl. Polym. Sci., 1988, 36, 635–643 CrossRef CAS.
- G. Pace, V. Ferri, C. Grave, M. Elbing, C. von Hänisch, M. Zharnikov, M. Mayor, M. A. Rampi and P. Samori, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9937–9942 CrossRef CAS.
- D. Yang, M. Piech, N. S. Bell, D. Gust, S. Vail, A. A. Garcia, J. Schneider, C. D. Park, M. A. Hayes and S. T. Picraux, Langmuir, 2007, 23, 10864–10872 CrossRef CAS.
- B. Bunker, D. Huber, J. Kushmerick, T. Dunbar, M. Kelly, C. Matzke, J. Cao, J. Jeppesen, O. J. Perkins, A. H. Flood and J. F. Stoddart, Langmuir, 2007, 23, 31–34 CrossRef CAS.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS.
- S. Patnaik, A. K. Sharma, B. S. Garg, R. P. Gandhi and K. C. Gupta, Int. J. Pharm., 2007, 342, 184–193 CrossRef CAS.
- X. Laloyaux and A. M. Jonas, Angew. Chem., Int. Ed., 2010, 49, 3262–3263 CrossRef CAS.
- G. Haberhauer and C. Kallweit, Angew. Chem., Int. Ed., 2010, 49, 2418–2421 CrossRef CAS.
- F. Schreiber, J. Phys.: Condens. Matter, 2004, 16, R881 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS.
- C. D. Bain, J. Evall and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 7155–7164 CrossRef CAS.
- R. G. Nuzzo, L. H. Dubois and D. L. Allara, J. Am. Chem. Soc., 1990, 112, 558–569 CrossRef CAS.
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 2000, 16, 7268–7274 CrossRef CAS.
- J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92–98 CrossRef CAS.
- K. Shin and E. J. Shin, Bull. Korean Chem. Soc., 2008, 29, 1259–1262 CrossRef CAS.
- S. Kucharski, R. Janik, H. Motschmann and C. Radüge, New J. Chem., 1999, 23, 765–771 RSC.
- M. Menzinger and R. L. Wolfgang, Angew. Chem., Int. Ed. Engl., 1969, 8, 438–444 CrossRef CAS.
- N. J. Dunn, W. H. Humphries IV, A. R. Offenbacher, T. L. King and J. A. Gray, J. Phys. Chem. A, 2009, 113, 13144–13151 CrossRef CAS.
- T. Kawai, J. Umemura and T. Takenaka, Langmuir, 1989, 5, 1378–1383 CrossRef CAS.
- G. Gauglitz and E. Scheerer, J. Photochem. Photobiol., A, 1993, 71, 205–212 CrossRef CAS.
- E. Vauthey, Chem. Phys., 1995, 196, 569–582 CrossRef CAS.
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188–8203 CrossRef CAS.
- H. Rau, Angew. Chem., Int. Ed. Engl., 1973, 12, 224–234 CrossRef.
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 1999, 15, 3759–3766 CrossRef CAS.
- C. Hoffmann and G. E. M. Tovar, J. Colloid Interface Sci., 2006, 295, 427–435 CrossRef CAS.
- B. Baisch, D. Raffa, U. Jung, O. M. Magnussen, C. Nicolas, J. Lacour, J. Kubitschke and R. Herges, J. Am. Chem. Soc., 2009, 131, 442–443 CrossRef CAS.
- C. Wöll, Angew. Chem., Int. Ed., 2009, 48, 8406–8408 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20151h/ |
| This journal is © The Royal Society of Chemistry 2012 |