Small-sized Ag nanocrystals: high yield synthesis in a solid–liquid phase system, growth mechanism and their successful application in the Sonogashira reaction†
Min
Han
,
Suli
Liu
,
Xiaopeng
Nie
,
Dan
Yuan
,
Peipei
Sun
,
Zhihui
Dai
* and
Jianchun
Bao
*
Jiangsu Key Laboratory of Biofunctional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, 210097, P. R. China. Tel: 0086 25 85891936E-mail: baojianchun@njnu.edu.cn; daizhihuii@njnu.edu.cn
First published on 1st May 2012
Abstract
In this paper, small-sized monodisperse Ag nanocrystals (NCs) have been successfully synthesized at gram-scale by thermal reduction of a large amount of solid AgNO3 (more than 10 mmol) with dodecylamine in 1-octadecene solvent. The formation process of the Ag NCs is different from that of a conventional homogeneous phase synthetic system. According to the temperature- and time-dependent experiments, a high temperature “digestive ripening” mechanism is suggested to elucidate their formation process. The size of Ag NCs can be easily controlled by the amount of solid AgNO3 added and the reaction temperature. Furthermore, the obtained Ag NCs are found to possess extraordinary catalytic activity, which can catalyze a series of Sonogashira reactions with high yield. Interestingly, under identical conditions, their catalytic activities are higher than that of similar sized Pd NCs, showing great promise for the substitution of conventional Pd-based catalysts to apply in the Sonogashira reaction. This developed synthetic strategy together with the fundamental understanding of heterogeneous nucleation and growth has great potential towards the contriving rational route for mass production of nanomaterials for advanced catalytic and other functional applications.
Introduction
Because of their broad use in catalysis, electronics, plasmonics, optical sensing, biological labeling, imaging, and photothermal therapy, the controlled synthesis of noble-metallic nanocrystals (NCs) with uniform geometries has been a research theme for decades.1–6 The properties of noble-metallic NCs are strongly determined by their size, shape, and surface structure. As an important noble metal, Ag NCs have attracted extensive attention. By far, many methods have been developed to synthesize Ag NCs, such as the polyol process,7 sonochemical method,8 photo-induced synthesis,9 biomolecule or polymer mediated chemical reduction,10 modification–dissolving–assembly strategy,11 seed-mediated growth,12 oil–water interface synthesis,13 and nonaqueous system thermal decomposition or reduction,14,15 and so on. Among those methods, the nonaqueous system thermal decomposition or reduction is one of the efficient strategies for preparing small-sized Ag NCs. For example, through thermal decomposition of Ag(CF3COO) in an oleic acid and bis-isoamyl ether system, monodisperse Ag NCs with sizes ranging from 7 to 11 nm were obtained.14 Also, by thermolysis of a Ag–oleate complex in the presence of oleic acid and dodecylamine, uniform Ag NCs with sizes less than 4 nm were synthesized.15 In those works, the Ag precursors have good solubilities in surfactant and solvent mixtures and the amount of used precursors was less than 10 mmol. Thus, the Ag precursors can be completely dissolved and the reaction systems are in a homogeneous phase. The dominant growth mechanism abides by the classical mode, i.e. burst nucleation and burst growth.16 If the Ag precursors have poor solubilities and the amount of them exceeds 10 mmol, the reaction system becomes a heterogeneous phase (solid–liquid phase). By far, there is only a few reports on the synthesis of Ag NCs through a solid–liquid phase system.17 In addition, for Ag NCs to be used in a wide range of practical applications, an economical mass production method with controllable growth needs to be developed. However, in most of the syntheses that have been reported so far, only sub-gram quantities of Ag NCs were synthesized. Therefore, developing simple routes for mass production of Ag NCs with uniform geometries remain a challenging task.On the other hand, as a class of important organic reactions, the Sonogashira reaction has always aroused the interest of chemists because it is used to synthesize natural products, bioactive compounds and electrooptical materials.18 To date, a few metal nanostructures such as Pd,19 Pd-based bimetal,20 Au,21 and ultrathin Ni22 have been used to catalyze this reaction, and Pd exhibits excellent catalytic performance. Due to the high cost of Pd metal, it is of great importance to replace it with a cheaper material. Although the unique optical and catalytic properties of Ag nanostructures have been extensively investigated up to now, however, there is no report using Ag NCs to catalyze the Sonogashira reaction. Therefore, exploring the catalytic application of Ag NCs in the Sonogashira reaction is an interesting topic.
Here, we report the synthesis of small-sized monodisperse Ag NCs via a solid–liquid phase chemical route. A large amount of solid AgNO3 (more than 10 mmol) is employed as the reactant and dodecylamine (DDA) is served as both capping and reducing reagent in 1-octadecene (ODE) solvent. By controlling the amount of solid AgNO3 and reaction temperature, monodisperse Ag NCs with sizes of 6, 9, and 11.5 nm can be easily obtained at a gram-scale. Based on the temperature- and time-dependent experimental results, a high temperature “digestive ripening” mechanism is suggested to explain the formation of Ag NCs. Furthermore, the Ag NCs are tested as catalysts for Sonogashira reactions. It is found that the obtained 6 nm Ag NCs exhibit extraordinary catalytic performance in the Sonogashira reaction with high yield. Interestingly, under the identical conditions, the catalytic activity of Ag NCs is higher than that of Pd NCs, showing great promise for the substitution of Pd-based catalysts for application in carbon–carbon cross-coupling reactions.
Experimental
Reagents
AgNO3 (> 99.8%), CuI, PPh3 and absolute alcohol were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). The DDA (> 98%), ODE (Tech, > 90%), and triphenylphosphine (PPh3) were purchased from Aldrich. The substrates used for catalysis, such as phenylacetylene, p-iodotoluene, and other terminal alkynes and aryl halides were purchased from Aldrich. Organic solvents including tetrahydrofuran (THF), N-methylpyrrolidone (NMP), N,N-dimethylformamide (DMF), acetonitrile, and isopropanol were distilled before use.Synthesis of Ag NCs
In a typical synthesis, a solid sample of AgNO3 (2.5–2.8 g), 1-octadecene (10 mL), and dodecylamine (8–10 mL) were added to a 250 mL three-neck flask at room temperature. Subsequently, the reactor was heated to 280–320 °C at a rate of 7 °C min−1 and kept at that temperature for 30 min. Finally, the reactor was naturally cooled down to 30 °C. The crude product was separated and re-dispersed in heptane. After adding absolute ethanol (50 mL) into the dispersed solution a large amount of precipitates were produced, which were separated by centrifugation and washed with heptane and absolute ethanol 2–3 times to remove by-products. Finally, the precipitate was dried in vacuum for 4 h and used for further characterization and analysis. The yield of Ag NCs was about 90%.The synthetic procedure of Pd NCs was similar to that of Ag NCs, which is given in ESI.†
Catalytic experiments
The catalytic synthesis of aryl alkynes by the obtained small-sized Ag NCs was as follows. 2 mL of NMP, 1 mmol of aryl halide, and 1 mmol of terminal alkynes were added in a clean and dry round flask, then 0.05 mmol of Ag NC catalyst, 0.05 mmol of PPh3, 0.04 mmol of CuI and 1.5 mmol of K2CO3 were added in turn. The mixture was stirred at 80 °C under a nitrogen atmosphere for the appropriate time (monitored by TLC) till the reaction was complete, then centrifuged. The solution was separated and the precipitate was washed with ether (3 × 5 mL). The solutions were combined and extracted with ether. Finally, they were purified by column chromatography on silica gel with hexane–ethyl acetate (volume ratio = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent to obtain the catalytic product. The Ag NC catalyst was recovered by further washing the precipitate with alcohol several times and then dried.
:1) as the eluent to obtain the catalytic product. The Ag NC catalyst was recovered by further washing the precipitate with alcohol several times and then dried.
Characterization
The X-ray diffraction (XRD) patterns were recorded on a powder sample using a D/max 2500 VL/PC diffractometer (Japan) equipped with graphite monochromatized Cu-Kα radiation (λ = 1.54060 Å) in 2θ ranging from 10 to 80 °C. Corresponding work voltage and current is 40 kV and 100 mA, respectively. MDI Jade 5.0 software was used to deal with the acquired diffraction data. The TEM images were taken on a JEM-200CX instrument (Japan), using an accelerating voltage of 200 kV. The HRTEM images were recorded on JEOL-2100 apparatus at an accelerating voltage of 200 kV. FE-SEM images were taken on a LEO-1530VP scanning electron microscope, operating at an accelerating voltage of 20 kV. The thermogravimetric analysis (TG) was performed on a PERKIN-ELMER TG/DTA synchronous thermoanalysis instrument. The X-ray energy dispersive spectra (EDS) were taken on a JSM-5610LV-Vantage typed energy spectrometer. The Sonogashira reaction products were identified by 1H-NMR, FT-IR and mass spectra. The 1H-NMR spectra were determined on a Bruker spectrometer (400 MHz) with TMS as the internal standard. FT-IR spectra were obtained with a Nexus 670 spectrometer. The mass spectra were performed on a Cary GC-MS-3800/2200 apparatus. The detailed 1H-NMR, FT-IR and mass spectra data of the Sonogashira reaction products were given in the ESI.†Results and discussion
TEM, HRTEM, XRD and TG analysis of monodisperse Ag NCs
The morphology and microstructure of the Ag NCs synthesized at 280 °C are examined by transmission electron microscopy (TEM). The low magnification TEM image in Fig. 1a shows large-area monodisperse quasi-spherical Ag NCs, which self-assemble into a 2D ordered array on a carbon-supported copper grid. The corresponding high magnification TEM image shown in Fig. 1b reveals that the average size of the Ag NCs is about 6 nm. The interspace between the neighboring NCs is about 2.4–3.0 nm. This is close to 2 times of the chain length of DDA, indicating that the Ag NCs are capped by DDA. HRTEM analysis (Fig. 1c) shows clear and continuous lattice fringes. The lattice spacing is measured to be 2.35 Å, corresponding to the interplanar separation between the (111) lattice plane of the face-centered cubic (fcc) phase of Ag. The XRD analysis (Fig. 1d) shows four distinct diffraction peaks, which can be indexed as (111), (200), (220) and (311) planes of fcc Ag (JCPDS No.04-0783). Both the HRTEM and XRD results reveal that the obtained Ag NCs are well crystallized. The thermogravimetric analysis demonstrates that the weight percentage of surface ligands adsorbed on the Ag NCs is about 6.3% (see ESI,† Fig. S1).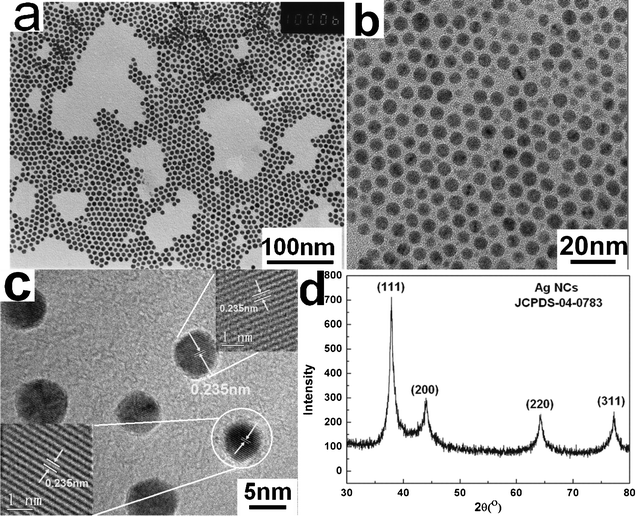 | ||
| Fig. 1 TEM images and XRD pattern of the Ag NCs synthesized at 280 °C. (a) Low and (b) high magnification TEM images of Ag NCs. (c) HRTEM image of Ag NCs. The inset shows the magnified lattice fringes. (d) XRD pattern of Ag NCs. | ||
Growth mechanism of monodisperse Ag NCs
The classical mode (burst nucleation and burst growth) is widely used to explain the formation of monodisperse metal and semiconductor NCs in the homogeneous phase synthetic system.16 However, this mode can't be adopted to elucidate the formation of monodisperse Ag NCs in our work. The reason is that the large amount of used AgNO3 which has a poor solubility in the mixed liquid of DDA and ODE causes the solid–liquid phase. In such a heterogeneous phase system, there is a difference on thermal transport between solid and liquid.23 Under temperature programming reaction mode, the solid AgNO3 is gradually reduced by DDA to form crystal nuclei with the increase of reaction temperature. Because the undissolved solid precursor provides nucleation centers or sites, the nucleation barrier in the reaction system is greatly reduced compared with that of the homogeneous phase system, causing the nucleation and a fast rate. A thin layer of black substance, metallic Ag, observed on the solid–liquid interface at 85 °C gives the direct evidence. Further increasing the reaction temperature, the solid AgNO3 is continuously reduced by DDA, which provides Ag sources for the growth of crystal nuclei. Because the metal NCs have a tendency for self-catalytic growth,24 the growth rate in the system may be fast and large multiply twinned or polyhedral Ag NCs are expected to be formed at low temperature. The TEM analysis of the samples taken at 130 and 160 °C confirms this speculation, as shown in Fig. 2a and 2b. A similar phenomenon has been observed in the synthesis of semiconductor NC assemblies by a using solid–liquid phase chemical route.25 As is well known, the in situ formed multiply twinned or polyhedral Ag NCs are kinetically stable.26 When the reaction temperature is further raised, they may be etched by oxidative species such as NO3− and the oxygen in air, leading to the evolution of their size and shape. Strong evidence comes from the experimental analysis of the samples taken at 200 and 260 °C. From the TEM images given in Fig. 2c and 2d, we can see that the initially formed large multiply twinned or polyhedral Ag NCs are gradually evolved into small quasi-spherical Ag NCs at temperatures ranging from 200 to 260 °C. These results demonstrate that the small-sized monodisperse Ag NCs are formed at a high temperature ripening stage. | ||
| Fig. 2 TEM images of the Ag NC samples taken at (a) 130, (b) 160, (c) 200, and (d) 260 °C, respectively. | ||
Generally, two ripening mechanisms are used to explain the “size focusing” process. One is Ostwald ripening,27 and the other is “digestive ripening”.28–30 We note that Chen et al. report the synthesis of nearly monodisperse Ag nanoparticles by reduction of a small amount of AgNO3 (150 mg) in an oleylamine–liquid paraffin system at 180 °C under a N2 atmosphere. They believe the ripening mechanism is “Ostwald ripening”.31 In order to understand the ripening mechanism of Ag NCs in our work, time-dependent experiments are performed to examine the ripening process at 280 °C. Four samples taken after ripening for 5, 10, 15, and 25 min are analyzed by TEM, as shown in Fig. 3. From Fig. 3a, we can see that except for a few large polyhedral Ag NCs (∼14.5–36 nm), plenty of quasi-spherical Ag NCs with the average size of ∼10 nm are obtained after ripening for 5 min. While ripening for 10 min, the large polyhedral Ag nanoparticles nearly disappear and many Ag nanospheres with the average size of ∼12.5 nm are formed (Fig. 3b). This phenomenon results from the dissolution of large polyhedral Ag nanoparticles, which provides Ag as the source for the growth of small Ag NCs. It indicates that the ripening mechanism is “digestive ripening”,28–30 different from the phenomenon observed by Chen et al.31 The origin of “digestive ripening” is usually attributed to the presence of excess surfactant.29 In our work, the used surfactant (DDA) is in excess, facilitating the occurrence of “digestive ripening”. However, this is not the sole origin based on the following experiment result by further prolonging the ripening time. It is found that the average size of Ag NCs reduces to 11.0 nm (Fig. 3c) after ripening for 15 min. The main origin of this size decrease phenomenon is attributed to the oxidative etching by prolonging ripening time. Similar oxidative etching has been observed by Kim et al. in the size-controllable synthesis of Ag nanoplates using KMnO4 as an oxidant.32 In our experiments, NO3− as well as oxygen in air (because the reaction system is open to air) promotes the etching process at high temperature. The further evidence comes from the experimental result observed at ripening for 25 min. Besides the formation of many 9.2 nm Ag NCs, some smaller NCs with sizes of ∼3–4 nm are also observed (Fig. 3d), indicating that the etching process has a serious effect on the size of Ag NCs in this case. Further prolonging the time to 30 min, the monodisperse Ag NCs with a size of ∼6 nm are generated (see Fig. 1). Based on the above analysis, the formation of small-sized monodisperse Ag NCs in our work is ascribed to the “digestive ripening” which results from the presence of excess DDA and oxidative etching at high temperature.
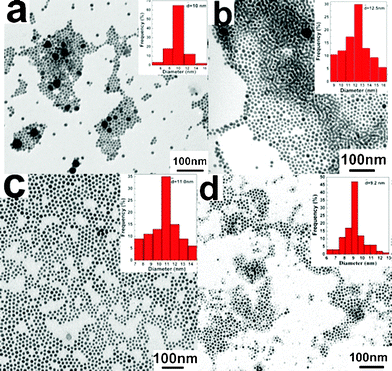 | ||
| Fig. 3 TEM images of the Ag NCs samples obtained at 280 °C by ripening for (a) 5 min, (b) 10 min, (c) 15 min, and (d) 25 min, respectively. (Notes: In the upper right corner of (a), only the statistical size of the small particles is listed. While in the upper right corner of (d), only the statistical size of the visible large particles is given. For observing clearly, the magnified TEM images are given in the ESI.†) | ||
Furthermore, in order to learn the feasibility of our proposed mechanism, some control experiments are performed. The results demonstrate that DDA plays an important role in the formation of small-sized monodisperse quasi-spherical Ag NCs.
If it is replaced by sodium dodecylsulfate (SDS), only large anisotropic and complicated one-dimensional Ag nanostructures are obtained (ESI,† Fig. S2). The reasons are as follows: (i) when DDA is replaced by SDS, there is no reducing reagent in the reaction system. Under this case, the formation of metallic Ag originates from the decomposition of solid AgNO3 at high temperature, not a reduction process like that by using DDA; (ii) SDS has different coordination ability from DDA, which may effectively prevent Ag NCs from being etched by oxidative species. Except for the “bifunctional” surfactant (DDA), it is also found that the amount of solid precursor and reaction temperature have an impact on the final size of the monodisperse Ag NCs. By increasing the amount of AgNO3 and raising the reaction temperature to 300 and 320 °C, monodisperse Ag NCs with sizes of ∼9 nm (Fig. 4a) and ∼11.5 nm (Fig. 4b) are also obtained, respectively. The corresponding XRD results are shown in Fig. 4c and 4d.
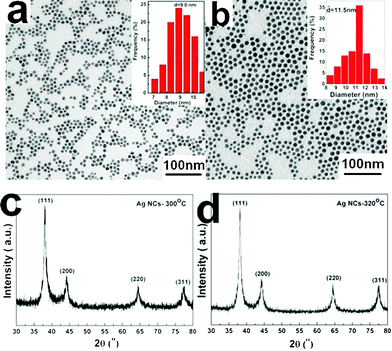 | ||
| Fig. 4 The TEM images (a, b) and XRD patterns (c, d) of the Ag NCs synthesized at 300 (a, c) and 320 °C (b, d). | ||
Catalytic properties
As an important noble metal, Ag NCs have been extensively used to catalyze many reactions, such as epoxidation of alkenes,33 partial oxidation of benzyl alcohols,34 oxidative coupling of methane,35 hydrogenation of nitrobenzene,36,37 electrocatalytic reduction of O2,38 and three-component coupling of aldehyde, alkyne and amine.39 However, to the best of our knowledge, Ag NC catalysts have not been used in the Sonogashira reaction. In the present work, the obtained Ag NCs with a size of 6, 9, and 11.5 nm are directly used as catalysts to catalyze the Sonogashira reaction without any surface treatment (Scheme 1). The experiments are performed at 80 °C under nitrogen atmosphere to avoid the oxidative coupling of terminal alkynes.40 The other reaction conditions are optimized by choosing the coupling reaction between phenylacetylene and p-iodotoluene as a model reaction. The influence factors, including the solvent, the amount of Ag NCs, the amount of CuI and PPh3, and the kind of base, are carefully examined. In pursuit of green catalysis, NMP is proven to be the best solvent for this reaction (ESI,† Table S1). The optimum amount of Ag NCs and PPh3 is 5 mol% (ESI,† Table S2 and S3). In order to inhibit the homocoupling reaction of terminal alkynes, the optimal amount of CuI is 4 mol% (ESI,† Table S4). Among the examined four kinds of inorganic bases (KOH, K2CO3, NaOH, and Na2CO3), K2CO3 gives the best result (ESI,† Table S5) and its optimal amount is about 1.5 equiv.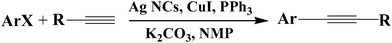 | ||
| Scheme 1 The chemical formula for the coupling of aryl halides with terminal alkynes by using Ag NCs as the catalyst. | ||
Under the optimized reaction conditions, the 6 nm Ag NCs are found to possess better catalytic performance (94% yield for the model reaction) than that of 9 nm (71%) and 11.5 nm Ag NCs (49%), revealing the size-dependent effect. The results of the other substrates used in the Sonogashira cross-coupling reaction catalyzed by 6 nm Ag NCs catalyst are summarized in Table 1. We find that both aryl iodides and bromides can react with aromatic or aliphatic terminal alkynes to give the corresponding cross-coupling products with high yields at 80 °C for less than 5 h. Also, the activity of p-iodotoluene is higher than that of m-iodotoluene and o-iodotoluene, showing an obvious stereospecific blockade effect of the aryl halides. In addition, the results reveal that the aryl halides with electron-withdrawing or electron-donating groups have no obvious influence on the catalytic reaction, implying that the reaction is relatively insensitive to the electronic characteristics of the substituent groups. The experimental results also reveal that the catalyst can be recycled and reused three times (see Table 1). After the three recycles, the activity of the catalyst is reduced. In order to tentatively understand the origin of deactivation, the catalyst is isolated from the organic layer after completion of the third recycle and characterized by EDX, XRD and TEM (ESI,† Fig. S3). The EDX and XRD results reveal that the component of the catalyst does not change after the catalytic reaction. No AgI product is detected, which is different from the previous report.41 While TEM analysis shows that there is a change of size and shape of Ag NCs, which may be the origin for the decrease of catalytic activity after being reused three times. In addition, the leaching and corresponding control experiments (see ESI†) show that the leaching amount of Ag NCs is about 2.04% in the reaction process and that the real catalyst is the Ag NC.
| Entry | ArX | R | Time (h) | Product | Yielda (%) |
|---|---|---|---|---|---|
| a Isolated yields. The products were characterized by 1H-NMR, FT-IR and Mass spectra. Corresponding data are given in electronic supplementary information (ESI). b The catalyst was reused for three times. | |||||
| 1 | C6H5I | CH3(CH2)3 | 2 | 1a13,15 | 93 |
| 2 | C6H5I | C6H5 | 2 | 1b14,16 | 89 |
| 3 | p-CH3C6H4I | CH3(CH2)3 | 2 | 1c | 91 |
| 4 | p-CH3C6H4I | C6H5 | 2 | 1d | 94 (93, 94, 91)b |
| 5 | m-CH3C6H4I | CH3(CH2)3 | 4 | 1e | 79 |
| 6 | m-CH3C6H4I | C6H5 | 3 | 1f | 86 |
| 7 | o-CH3C6H4I | CH3(CH2)3 | 5 | 1g | 74 |
| 8 | o-CH3C6H4I | C6H5 | 5 | 1h | 83 |
| 9 | p-Cl-C6H4I | CH3(CH2)3 | 2 | 1i | 89 |
| 10 | p-Cl-C6H4I | C6H5 | 2 | 1j | 94 |
| 11 | p-OCH3C6H4Br | CH3(CH2)3 | 5 | 1k | 88 |
| 12 | p-OCH3C6H4Br | C6H5 | 4 | 1l | 91 |
| 13 | C6H5Br | CH3(CH2)3 | 5 | 1a | 87 |
| 14 | C6H5Br | C6H5 | 5 | 1b | 82 |
| 15 | C6H5Cl | CH3(CH2)3 | 24 | 1a | Trace |
| 16 | C6H5Cl | C6H5 | 24 | 1b | Trace |
For comparing the catalytic performance of Ag NCs with that of Pd NCs, we use Pd(NO3)2 instead of AgNO3 and try to synthesize similar sized Pd NCs under the same synthetic system. Detailed structural characterization of the Pd NCs is given in the ESI,† Fig. S4. The cross-coupling reaction of p-iodotoluene with phenylacetylene is selected as the criteria for comparison. The amount of solvent, catalyst, co-catalyst, ligand, and base are identical to that used for Ag NCs. The results reveal that both the reactions are completed at 80 °C for 2 h by using Pd NCs and Ag NCs as the catalysts. For Pd NCs, the isolated yield is 90%. While for Ag NCs, the isolated yield is 94%. Because some surface ligands are adsorbed on Ag NCs and Pd NCs, the actual and effective catalytic active surface areas are difficult to measure. So, the turnover frequency (TOF) values for Ag NCs and Pd NCs catalysts can't be exactly calculated. But the turnover mole number of substrates (average value) based on unit mass catalyst under unit time (abbrev. TMT) of Ag NCs and Pd NCs can be exactly calculated. The calculated TMT values for Ag NCs and Pd NCs catalysts are 87.1 mmol g−1catalyst h−1 and 84.6 mmol g−1catalyst h−1, respectively. In order to get further insight on the catalytic performance differences of Ag NCs and Pd NCs, catalytic dynamic experiments are carried out. Corresponding kinetic plots for Ag NCs and Pd NCs are shown in ESI,† Fig. S5. From those plots, we can see that the reaction times for Ag NCs and Pd NCs to reach 50% conversion (abbrev. T50%) are 54 and 64 min, respectively. Based on both TMT and T50% values, the catalytic activity of Ag NCs is higher than that of Pd NCs. The different activities of Ag NCs and Pd NCs may be mainly attributed to the differences in electronic configurations of Ag ([Kr] 4d105s1) and Pd ([Kr]4d10). The 5s orbital of Ag has an unpaired electron while that of Pd has no isolated electrons. In contrast to Pd, Ag can easily lose the outermost layer 5s electron to form Ag+, which is beneficial to the key oxidative addition step in the Sonogashira reaction.18 In addition, it should be mentioned that our Ag NC catalyst possesses a compatible or even higher catalytic activity than those of previously reported Pd nanoparticles and Pd-based bimetallic nanoparticles catalysts.19,20 These results show that Ag NCs can be served as an ideal candidate catalyst for the substitution of Pd-based catalysts to apply in the Sonogashira reaction.
Conclusions
In summary, monodisperse Ag NCs with sizes of about 6, 9, and 11.5 nm have been successfully prepared at a gram-scale via a simple and mild solid–liquid phase chemical route under temperature programming reaction mode. A series of experimental results show that the growth process of Ag NCs in the solid–liquid phase system is different from that of a conventional homogeneous phase synthetic system. The size of Ag NCs can be easily adjusted by the amount of solid AgNO3 added and the reaction temperature. Based on the temperature- and time-dependent experimental results, a high temperature “digestive ripening” mechanism is supposed to explain their formation process. The obtained Ag NCs can be directly used as catalysts in Sonogashira reactions, exhibiting size-dependent catalytic properties. The 6 nm Ag NCs are found to possess remarkable catalytic performance with high yield. Also, the Ag NCs catalyst can be recycled and reused three times without loss of catalytic activity. Interestingly, under the identical conditions, Ag NCs exhibit higher activity than Pd NCs with a similar size, showing great promise for substitution of conventional Pd-based catalysts to be applied in the Sonogashira reaction. This work not only provides a developed synthetic strategy for mass production of small-sized metal NCs but also finds an ideal candidate catalyst for applying in the important C–C cross-coupling reaction.Acknowledgements
This work was supported by the National Natural Science Foundation of China under research project (20901041, 21175069, and 21171096), the program for New Century Excellent Talents in University of Chinese Ministry of Education (NCET-09-0159), Foundation of the Jiangsu Education Committee (11KJA150003) and the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Program for Outstanding Innovation Research Team of Universities in Jiangsu Province.References
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS.
- (a) V. Mazumder and S. H. Sun, J. Am. Chem. Soc., 2009, 131, 4588 CrossRef CAS; (b) J. Kim, Y. Lee and S. H. Sun, J. Am. Chem. Soc., 2010, 132, 4996 CrossRef CAS; (c) O. Metin, V. Mazumder, S. Ozkar and S. H. Sun, J. Am. Chem. Soc., 2010, 132, 1468 CrossRef CAS; (d) J. B. Wu, J. L. Zhang, Z. M. Peng, S. C. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984 CrossRef CAS.
- (a) H. J. Lee, S. E. Habas, G. A. Somorjai and P. D. Yang, J. Am. Chem. Soc., 2008, 130, 5406 CrossRef CAS; (b) M. S. Jin, H. Y. Liu, H. Zhang, Z. X. Xie, J. Y. Liu and Y. N. Xia, Nano Res., 2011, 4, 83 CrossRef CAS; (c) K. P. Gong, D. Su and R. R. Adzic, J. Am. Chem. Soc., 2010, 132, 14364 CrossRef CAS; (d) S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333 CrossRef CAS.
- (a) X. Q. Huang, S. H. Tang, X. L. Mu, Y. Dai, G. X. Chen, Z. Y. Zhou, F. X. Ruan, Z. L. Yang and N. F. Zheng, Nat. Nanotechnol., 2011, 6, 28 CrossRef CAS; (b) F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, F. Aksoy, S. Aloni, V. Altoe, S. Alayoglu, J. R. Renzas, C. K. Tsung, Z. W. Zhou, Z. Liu, M. Salmeron and G. A. Sormojai, J. Am. Chem. Soc., 2010, 132, 8697 CrossRef CAS; (c) N. Gupta, H. P. Singh and R. K. Sharma, J. Mol. Catal. A: Chem., 2011, 335, 248 CrossRef CAS; (d) S. L. Shen, J. Zhuang, Y. Yang and X. Wang, Nanoscale, 2011, 3, 272 RSC.
- (a) J. Park, E. Kang, S. U. Son, H. M. Park, M. K. Lee, J. Kim, K. W. Kim, H. J. Noh, J. H. Park, C. J. Bae, J. G. Park and T. Hyeon, Adv. Mater., 2005, 17, 429 CrossRef CAS; (b) M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffman, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617 CrossRef CAS; (c) Y. H. Chen, H. H. Huang and M. H. Huang, J. Am. Chem. Soc., 2009, 131, 9114 CrossRef CAS.
- (a) D. S. Wang and Y. D. Li, Adv. Mater., 2011, 23, 1044 CrossRef CAS; (b) Y. E. Wu, D. S. Wang, P. Zhao, Z. Q. Niu, Q. Peng and Y. D. Li, Inorg. Chem., 2011, 50, 2046 CrossRef CAS; (c) I. Nakamula, Y. Yamanoi, T. Imaoka, K. Yamamoto and H. Nishihara, Angew. Chem., Int. Ed., 2011, 50, 5830 CrossRef CAS.
- (a) B. Wiley, Y. G. Sun and Y. N. Xia, Acc. Chem. Res., 2007, 40, 1067 CrossRef CAS; (b) X. Liu, R. Huang and J. Zhu, Chem. Mater., 2008, 20, 192 CrossRef CAS.
- F. Tao, Z. J. Wang, D. B. Chen, L. Z. Yao, W. L. Cai and X. G. Li, Nanotechnology, 2007, 18, 295602 CrossRef.
- (a) R. Jin, Y. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901 CrossRef CAS; (b) P. D. Cozzoli, R. Compareli, E. Fanizza, M. L. Curri, A. Agostiano and D. Laub, J. Am. Chem. Soc., 2004, 126, 3868 CrossRef CAS; (c) L. Maretti, P. S. Billone, Y. Liu and J. C. Scaiano, J. Am. Chem. Soc., 2009, 131, 13972 CrossRef CAS.
- (a) H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. Labean, Science, 2003, 301, 1882 CrossRef CAS; (b) M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS; (c) L. T. Yu, I. A. Banerjee and H. Matsui, J. Am. Chem. Soc., 2003, 125, 14837 CrossRef CAS; (d) T. H. Lee, J. I. Gonzalez, J. Zhang and R. M. Dickson, Acc. Chem. Res., 2005, 38, 534 CrossRef CAS; (e) J. Bai, Y. Qin, C. Jiang and L. Qi, Chem. Mater., 2007, 19, 3367 CrossRef CAS; (f) P. Gao, C. L. Zhan and M. H. Liu, Langmuir, 2006, 22, 775 CrossRef CAS; (g) Z. Shen, H. W. Duan and H. Frey, Adv. Mater., 2007, 19, 349 CrossRef CAS.
- Y. H. Wang, P. L. Chen and M. H. Liu, Nanotechnology, 2008, 19, 045607 CrossRef.
- (a) N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Commun., 2001, 617 RSC; (b) Q. Zhang, W. Y. Li, C. Moran, J. Zeng, J. Y. Chen, L. P. Wen and Y. N. Xia, J. Am. Chem. Soc., 2010, 132, 11372 CrossRef CAS.
- (a) C. N. R. Rao and K. P. Kalyanikutty, Acc. Chem. Res., 2008, 41, 489 CrossRef CAS; (b) H. Y. Shi, B. Hu, X. C. Yu, R. L. Zhao, X. F. Ren, S. L. Liu, J. W. Liu, M. Feng, A. W. Xu and S. H. Yu, Adv. Funct. Mater., 2010, 20, 958 CrossRef CAS.
- X. Z. Lin, X. Teng and H. Yang, Langmuir, 2003, 19, 10081 CrossRef CAS.
- P. Li, Q. Peng and Y. D. Li, Chem.–Eur. J., 2011, 17, 941 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630 CrossRef CAS.
- (a) X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121 CrossRef CAS; (b) M. Yamamoto and M. Nakamoto, J. Mater. Chem., 2003, 13, 2064 RSC.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS.
- (a) A. R. Gholap, K. Venkatesan, R. Pasricha, T. Daniel, R. J. Lahoti and K. V. Srinivasan, J. Org. Chem., 2005, 70, 4869 CrossRef CAS; (b) S. Y. Gao, H. J. Zhang, X. M. Wang, W. P. Mai, C. Y. Peng and L. H. Ge, Nanotechnology, 2005, 16, 1234 CrossRef CAS; (c) C. H. Xue, K. Palaniappan, G. Arumugam, S. A. Hackney, J. Liu and H. Y. Liu, Catal. Lett., 2007, 116, 94 CrossRef CAS; (d) J. Z. Jiang, Y. A. Wei and C. Cai, J. Colloid Interface Sci., 2007, 312, 439 CrossRef CAS; (e) L. P. Chen, S. G. Hong, X. P. Zhou and H. Q. Hou, Catal. Commun., 2008, 9, 2221 CrossRef CAS; (f) S. Sawoo, D. Srimani, P. Dutta, R. Lahiri and A. Sarkar, Tetrahedron, 2009, 65, 4367 CrossRef CAS.
- (a) S. U. Son, Y. Jang, J. Park, H. B. Na, H. M. Park, H. J. Yun, J. Lee and T. Hyeon, J. Am. Chem. Soc., 2004, 126, 5026 CrossRef CAS; (b) Y. Li, P. Zhou, Z. H. Dai, Z. Hu, P. P. Sun and J. C. Bao, New J. Chem., 2006, 30, 832 RSC; (c) H. Li, Z. H. Zhu, J. Liu, S. H. Xie and H. X. Li, J. Mater. Chem., 2010, 20, 4366 RSC; (d) H. Li, Z. H. Zhu, H. X. Li, P. Li and X. G. Zhu, J. Colloid Interface Sci., 2010, 349, 613 CrossRef CAS; (e) P. Venkatesan and J. Santhanalakshmi, Langmuir, 2010, 26, 12225 CrossRef CAS; (f) J. Guerra and M. A. Herrero, Nanoscale, 2010, 2, 1390 RSC.
- (a) C. González-Arellano, A. Abad, A. Corma, H. García, M. Iglesias and F. Sánchez, Angew. Chem., Int. Ed., 2007, 46, 1536 CrossRef; (b) S. K. Beaumont, G. Kyriakou and R. M. Lambert, J. Am. Chem. Soc., 2011, 132, 12246 CrossRef.
- L. Wang, P. H. Li and Y. C. Zhang, Chem. Commun., 2004, 514 RSC.
- M. Han, N. E. Shi, W. L. Zhang, B. J. Li, J. H. Sun, K. J. Chen, J. M. Zhu, X. Wang and Z. Xu, Chem.–Eur. J., 2008, 14, 1615 CrossRef CAS.
- C. Besson, E. E. Finney and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 8179 CrossRef CAS.
- M. Han, Y. R. Li, H. Y. Niu, L. L. Liu, K. J. Chen, J. C. Bao, Z. H. Dai and J. M. Zhu, Chem.–Eur. J., 2011, 17, 3739 CrossRef CAS.
- S. Peng and Y. G. Sun, Chem. Mater., 2010, 22, 6272 CrossRef CAS.
- (a) T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and A. A. EI-Sayed, Science, 1996, 272, 1924 CAS; (b) C. L. Jiang, W. Q. Zhang, Y. K. Liu and Y. T. Qian, Cryst. Growth Des., 2006, 6, 2603 CrossRef CAS.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen and K. J. Klabunde, Chem. Mater., 2003, 15, 935 CrossRef CAS.
- A. C. S. Samia, K. Hyzer, J. A. Schlueter, C. L. Qin, J. S. Jiang, S. D. Bader and X. M. Lin, J. Am. Chem. Soc., 2005, 127, 4126 CrossRef CAS.
- A. C. S. Samia, J. A. Schlueter, J. S. Jiang, S. D. Bader, C. J. Qin and X. M. Lin, Chem. Mater., 2006, 18, 5203 CrossRef CAS.
- M. Chen, Y. G. Feng, X. Wang, T. C. Li, J. Y. Zhang and D. J. Qian, Langmuir, 2007, 23, 5296 CrossRef CAS.
- J. Roh, J. Yi and Y. Kim, Langmuir, 2010, 26, 11621 CrossRef CAS.
- T. M. Bernhardt, Int. J. Mass Spectrom., 2005, 243, 1 CrossRef CAS.
- R. Yamamoto, Y. Sawayama, H. Shibahara, Y. Ichihashi, S. Nishiyama and S. Tsuruya, J. Catal., 2005, 234, 308 CrossRef CAS.
- A. J. Nagy, G. Mestl and R. Schlögl, J. Catal., 1999, 188, 58 CrossRef CAS.
- Y. Y. Chen, C. Wang, H. Y. Liu, J. S. Qiu and X. H. Bao, Chem. Commun., 2005, 5298 RSC.
- M. Han, X. Li, B. J. Li, K. J. Chen, Z. Xu and J. M. Zhu, J. Phys. Chem. C, 2008, 112, 17893 CAS.
- L. Kuai, B. Y. Geng, S. Z. Wang, Y. Y. Zhao, Y. C. Luo and H. Jiang, Chem.–Eur. J., 2011, 17, 3482 CrossRef CAS.
- W. J. Yan, R. Wang, Z. Q. Xu, J. K. Xu, L. Lin, Z. Q. Shen and Y. F. Zhou, J. Mol. Catal. A: Chem., 2006, 255, 81 CrossRef CAS.
- S. Thorand and N. Krause, J. Org. Chem., 1998, 63, 8551 CrossRef CAS.
- P. Li and L. Wang, Synlett, 2006, 14, 2261 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: the TG plot of Ag NCs, the FE-SEM images of Ag nanostructures in control experiments, experimental data for optimizing catalytic conditions, 1H NMR, FT-IR and mass spectra data of catalytic products, the structural characterization of control catalyst, the leaching and control experiments, and the catalytic dynamic plots of Ag NCs and Pd NCs. See DOI: 10.1039/c2ra20119d/ |
| This journal is © The Royal Society of Chemistry 2012 |