DOI:
10.1039/C2RA20111A
(Paper)
RSC Adv., 2012,
2, 6680-6685
Isostructural lanthanide oxalatophosphonates Ln(5pm8hqH3)(C2O4)1.5(H2O)·2H2O [Ln(III) = Eu, Gd, Tb, Dy] (5pm8hqH3 = 5-phosphonomethyl-8-hydroxyquinoline): structures, magnetic and fluorescent properties†
Received
19th January 2012
, Accepted 18th May 2012
First published on 21st May 2012
Abstract
Isostructural lanthanide oxalatophosphonates Ln(5pm8hqH3)(C2O4)1.5(H2O)·2H2O [Ln(III) = Eu(1), Gd(2), Tb(3), Dy(4)] have been obtained through hydrothermal reactions of 5-phosphonomethyl-8-hydroxyquinoline (5pm8hqH3), oxalate, and Ln(NO3)3·6H2O. Four compounds reveal layer structures, in which {LnO8} polyhedra are linked through oxalate units into a netlike {Ln(C2O4)1.5}n layer containing 24-member ring composed of six Ln(III) ions and six oxalate anions. The noncoordinated 8-hydroxyquinoline groups protrude from two sides of the layer. Neighboring layers are further connected to each other through hydrogen bonds and aromatic stacking of the 8-hydroxyquinoline rings, forming a supramolecular 3D structure. In magnetic measurements, compound 2 shows weak antiferromagnetic interactions. The magnetic behaviors of 1, 3 and 4 are ascribed to the depopulation of the Stark levels and weak antiferromagnetic interactions. In the solid-state fluorescent measurements, strong ligand-centered emissions and weak Ln(III)-centered emissions can be observed for 1 and 4, while only ligand-centered emissions for 2 and 3.
Introduction
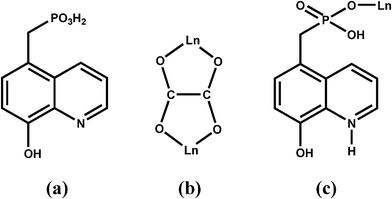
In recent years, considerable effort has been focused on lanthanide phosphonates due to their structural diversity and interesting luminescent1 and magnetic properties.2 In the preparation of lanthanide phosphonates, the introduction of oxalate is one of the effective approaches because of three main advantages of oxalate: (i) improving the crystallization of the products1b,3 (ii) mediating electronic effects between paramagnetic metal ions, and (iii) linking metal centers into various structures with dimensionalities ranging from zero to three.4 On the other hand, lanthanide phosphonates with structural diversity and related physical and chemical properties could be obtained by modifying phosphonates with different functional organic groups, such as crown ether,5 carboxylate,2c,6 amino,2c,7 and pyridyl,7b,8etc. 8-Hydroxyquinoline and its derivatives are versatile coordination ligands towards a wide range of metal ions, including Ln(III) ions, resulting in some materials with promising photophysical properties.9,10 The incorporation of the 8-hydroxyquinoline group into a phosphonate ligand could not only provide additional coordination sites for the metal ions but also influence the packing of the structures through weak interactions such as aromatic stacking and hydrogen-bond interactions. So far, only one case of phosphonate ligand containing 8-hydroxyquinoline group has been reported, i.e. 8-quinolinyloxymethylphosphonic acid, synthesized by phosphonomethylation of 8-hydroxyquinoline at the hydroxyl position.11 Based on the corresponding diethyl-8-quinolinyloxymethylphosphonate, zinc compound with chain structure was obtained. In order to obtained more lanthanide phosphonates with interesting structures and properties, we synthesized 5-phosphonomethyl-8-hydroxyquinolin (5pm8hqH3) (Scheme 1a). Based on 5pm8hqH3 and the second ligand of oxalate, isostructural lanthanide oxalatophosphonates Ln(5pm8hqH3)(C2O4)1.5(H2O)·2H2O [Ln(III) = Eu(1), Gd(2), Tb(3), Dy(4)] have been prepared. Their structures, magnetic and fluorescent properties were studied.
 |
| | Scheme 1 | |
Experimental
Materials and physical measurements
The 5-chloromethyl-8-hydroxyquinol hydrochloride was prepared according the literature.12 All the other starting materials were of reagent grade and were obtained from commercial sources without further purification. Elemental analyses were performed on a Perkin Elmer 240C elemental analyzer. IR spectra were obtained as KBr disks on a VECTOR 22 spectrometer. Thermal analyses were performed under N2 gas flow using a Mettler-Toloedo TGA/DSC instrument. The heating rate was 10 °C min−1 from room temperature to 800 °C. The powder XRD patterns were recorded on a Shimadzu XD-3A X-ray diffractometer. Magnetic susceptibility data were obtained on microcrystalline samples (6.01 mg for 1, 3.88 mg for 2, 3.96 mg for 3, and 4.59 mg for 4), using a Quantum Design MPMS-XL7 SQUID magnetometer. Diamagnetic corrections were made for both the sample holder and the compound estimated from Pascal's constants.13 The luminescent spectra were measured at room temperature on a Perkin Elmer LS55 fluorescence spectrometer. The luminescent lifetimes were measured at room temperature on an Edinburgh FL-FS920 fluorescence spectrometer.
Synthesis of 5-phosphonomethyl-8-hydroxyquinoline hydrochloride (5pm8hqH3·HCl·H2O)
5-Chloromethyl-8-hydroxyquinol hydrochloride (10 g, 40.8 mmol) was dissolved in 200 mL of deionized water, and saturated Na2CO3 aqueous solution was added under stirring until the pH was near to 7.0. The ashen solid was collected, washed with water, and dried in air. The mixture of ashen solid and P(OEt)3 (68 g, 408 mmol) was heated at 160 °C for 32 h. After removing unreacted P(OEt)3 by rotary evaporation, the resultant brown oil was mixed with 6M HCl (100 mL). The mixture was refluxed for 12 h, and then evaporated until yellow solid just appears. After cooling, yellow needlelike crystals were collected, washed with ethanol, and dried in air. Yield: 8.7 g (72.6%). Anal. found (calcd) for C10H13O5NPCl: C, 41.52 (40.90); H, 4.55 (4.46); N, 4.51 (4.77). IR (KBr, cm−1): 3410(s), 2950(s, br), 2665(m), 2098(w), 1628(m), 1597(s), 1553(s), 1508(w), 1472(w), 1421(w), 1390(s), 1306(s), 1250(m), 1203(w), 1158(w), 1088(s), 990(s), 923(s), 849(w), 829(m), 818(s), 779(w), 733(w), 703(w), 667(w), 637(w), 547(w), 515(w), 478(s), 415(m).
Syntheses of Ln(C2O4)1.5(5pm8hqH3)(H2O)·2H2O [Ln(III) = Eu(1), Gd(2), Tb(3), Dy(4)]
Compounds 1–4 were synthesized according to the same procedure. In the synthesis of 1, a mixture of Eu(NO3)3·6H2O (0.10 mmol, 0.0446 g), 5pm8hqH3·HCl·H2O (0.14 mmol, 0.0414 g), and Na2C2O4 (0.15 mmol, 0.0201 g) in 5 mL of water was kept in a Teflon-lined autoclave at 140 °C for one day. After cooling to around 90 °C, yellow blocky crystals were collected as a monophasic material based on the powder XRD patterns (Fig. S1†). Compounds 2–4 were synthesized using Gd(NO3)3·6H2O, Tb(NO3)3·6H2O and Dy(NO3)3·6H2O, respectively, instead of Eu(NO3)3·6H2O. The yields, elemental analyses, and IR data for compounds 1–4 were shown in the ESI.† Thermal analyses of compounds 1–4 revealed a weight loss of 9.6% in 46–235 °C for 1, 9.4% in 41–248 °C for 2, 9.1% in 37–260 °C for 3, and 9.3% in 47–280 °C for 4, respectively, close to the theoretical values of one coordination and two lattice water molecules (9.4% for 1, 9.3% for 2, 9.2% for 3, and 9.2% for 4 (Fig. S2†). In addition, TG curves of 1–4 indicate that the removal of coordination water molecules resulted in the structural decomposition of compounds, which was confirmed by XRD patterns.
X-ray crystallographic studies
The diffraction data of 1–4 were collected on a Bruker SMART APEX CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.710 73 Å) at room temperature. A hemisphere of data was collected using a narrow-frame method with scan widths of 0.30° in ω and an exposure time of 10 s per frame. The data were integrated using the Siemens SAINT program,14 with the intensities corrected for the Lorentz factor, polarization, air absorption, and absorption due to variation in the path length through the detector faceplate. Multi-scan absorption corrections were applied. The structures were solved by direct methods and refined on F2 by full matrix least squares using SHELXTL.15 All the non-hydrogen atoms were located from the Fourier maps, and were refined anisotropically. All H atoms were put in calculated positions using riding model, and were refined isotropically, with the isotropic vibration parameters related to the non-H atom to which they are bonded. The crystallographic data for 1–4 are listed in Table 1. Selected bond lengths are given in Table 2. CCDC 810422–810425 contain the supplementary crystallographic data for 1–4 for this paper.†
| Compound |
1
|
2
|
3
|
4
|
|
R
1 = Σ||Fo| − |Fc||/Σ|Fo|, wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2.
|
| Empirical formula |
C13H16O13NPEu |
C13H16O13NPGd |
C13H16O13NPTb |
C13H16O13NPDy |
| Fw |
577.20 |
582.49 |
584.16 |
587.74 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
|
a/Å |
7.864(3) |
7.8439(12) |
7.8354(13) |
7.8235(2) |
|
b/Å |
9.964(3) |
9.9333(15) |
9.9100(17) |
9.9093(2) |
|
c/Å |
12.842(4) |
12.839(2) |
12.852(2) |
12.840(3) |
|
α (°) |
67.521(5) |
67.578(3) |
67.780(3) |
67.621(1) |
|
β (°) |
89.856(6) |
89.872(3) |
89.721(3) |
89.760(1) |
|
γ (°) |
80.711(6) |
80.597(2) |
80.236(3) |
80.470(1) |
|
V Å3 |
915.6(5) |
910.3(2) |
908.5(3) |
905.88(4) |
|
Z
|
2 |
2 |
2 |
2 |
|
ρ
c/g cm−3 |
2.094 |
2.125 |
2.135 |
2.155 |
|
F(000) |
566 |
568 |
570 |
572 |
| Crystal size/mm |
0.09 × 0.07 × 0.06 |
0.10 × 0.08 × 0.07 |
0.10 × 0.08 × 0.06 |
0.11 × 0.09 × 0.08 |
| Theta range (°) |
1.72–25.00° |
2.25–24.99° |
2.25–24.99° |
2.25–25.25° |
| Reflections collected/unique |
4581, 3154 |
4587, 3144 |
4551, 3133 |
4887, 3240 |
| (Rint = 0.0181) |
(Rint = 0.0251) |
(Rint = 0.0277) |
(Rint = 0.0617) |
| GOF on F2 |
1.074 |
1.002 |
1.057 |
1.031 |
|
R
1, wR2a [I > 2σ(I)] |
0.0317, 0.0781 |
0.0288, 0.0450 |
0.0287, 0.0506 |
0.0269, 0.0580 |
|
R
1, wR2a (all data) |
0.0357, 0.0793 |
0.0340, 0.0454 |
0.0336, 0.0511 |
0.0338, 0.0597 |
| (Δρ)max, (Δρ)min/e Å−3 |
1.335, −0.627 |
0.788, −0.686 |
1.112, −0.684 |
0.855, −1.623 |
Table 2 Selected bond lengths (Å) for 1–4a
| |
1
|
2
|
3
|
4
|
|
Symmetry codes: A: −x + 1, −y + 1, −z + 1; B: −x + 1, −y + 2, −z + 1; C: −x + 2, −y + 1, −z + 1.
|
| Ln1–O1 |
2.250(4) |
2.235(3) |
2.223(3) |
2.209(3) |
| Ln1–O1W |
2.420(4) |
2.389(3) |
2.391(3) |
2.372(3) |
| Ln1–O9 |
2.417(3) |
2.411(3) |
2.397(3) |
2.385(3) |
| Ln1–O6A |
2.420(4) |
2.409(3) |
2.403(3) |
2.389(3) |
| Ln1–O10B |
2.423(3) |
2.403(3) |
2.387(3) |
2.402(3) |
| Ln1–O7 |
2.455(3) |
2.429(3) |
2.413(3) |
2.408(3) |
| Ln1–O8C |
2.429(3) |
2.415(3) |
2.404(3) |
2.404(3) |
| Ln1–O5 |
2.435(4) |
2.419(3) |
2.416(3) |
2.408(3) |
| P1–O1 |
1.501(4) |
1.500(3) |
1.506(3) |
1.500(3) |
| P1–O2 |
1.559(4) |
1.562(3) |
1.570(4) |
1.564(4) |
| P1–O3 |
1.504(4) |
1.506(3) |
1.504(4) |
1.505(3) |
| O4–C8 |
1.349(6) |
1.347(5) |
1.350(5) |
1.342(6) |
| O5–C11 |
1.242(6) |
1.243(5) |
1.244(6) |
1.240(5) |
| O6–C11 |
1.247(6) |
1.255(5) |
1.259(6) |
1.265(5) |
| O7–C12 |
1.249(6) |
1.261(5) |
1.261(6) |
1.267(5) |
| O8–C12 |
1.240(6) |
1.235(5) |
1.236(6) |
1.250(5) |
| O9–C13 |
1.246(6) |
1.240(5) |
1.232(6) |
1.248(5) |
| O10–C13 |
1.254(6) |
1.261(5) |
1.271(5) |
1.258(5) |
Results and discussion
Syntheses
Compounds 1–4 were prepared through hydrothermal reactions of 5pm8hqH3·HCl·H2O, Na2C2O4 and corresponding lanthanide nitrates at 140 °C for one day. It was found that the Ln/oxalate/ligand molar ratio could greatly affect the formation of 1–4. The molar ratio of 1/1.5/1 results in a mixture of product and some white powders, while the molar ratio of 1/1.5/1.4 leads to a mixture of product and a small amount of yellow needlelike crystals of phosphonate ligand. Finally, compounds 1–4, with the best crystal quality and relative high yield, were prepared by using the Ln/oxalate/ligand molar ratio of 1/1.5/1.4, and by filtrating product when the temperature of reaction mixture is about 90 °C to avoid the crystallization of excess phosphonate ligand in solution.
Crystal structures of 1–4
Compounds 1–4 are isostructural and crystallize in the triclinic space group P. As shown in Fig. 1, the asymmetric unit of 1 contains one Eu(III) ion, one 5pm8hqH3 ligand, 1.5 oxalate anions, one coordination and two lattice water molecules. The Eu(III) is eight-coordinated and is surrounded by six oxalate oxygen atoms (O5, O7, O9, O6A, O8C, O10B), one phosphonate oxygen (O1), and one water molecule (O1W). The Eu–O bond distances are in the range of 2.250(4)–2.455(3) Å, comparable to those in compounds [Eu2(Hpmp)(C2O4)2.5(H2O)3]·H2O (H2pmp = N-piperidinomethane-1-phosphonic acid) [2.258(4)–2.509(5) Å]16 and [Eu((HO3PCH2)2NHCH2C6H5)(C2O4)]·2H2O [2.324(3)–2.478(3) Å].17 Each oxalate anion adopts the most commonly observed bis-bidentate coordination mode (Scheme 1b), chelating to two Eu(III) ions. The connections of oxalate units and Eu(III) ions result in a lanthanide oxalate layer of {Ln(C2O4)1.5}n in approximate ab plane (Fig. 2a). This layer contains parallelogram-like 24-member ring made up of six Ln(III) ions and six oxalate units, showing the Ln⋯Ln separation of 6.255(1)–6.272(6) Å over the oxalate unit (Fig. 2b). The similar lanthanide oxalate layer was observed in compounds [Ln2(H4L)(C2O4)3(H2O)]3·2H2O [Ln = Gd, Tb, Dy, Y, Er, Lu; H4L = 3-pyridyl-CH2N(CH2PO3H2)2], where the {Ln2(C2O4)3}n layers are further interlinked by phosphonate groups through sharing Ln(III) ions to form three dimensional framework.18 In the 5pm8hqH3 ligand, only phosphonate oxygen O1 is involved in the coordination to one Eu(III) ion (Scheme 1c). The phosphonate oxygen atom O2, hydroxyl oxygen atom O4 and pyridine nitrogen atom N1 are protonated [P1–O2 = 1.559(4) Å, C8–O4 = 1.349(6) Å], while the remaining phosphonate oxygen O3 is pendant [P1–O3 = 1.504(4) Å]. Thus, 5pm8hqH3 molecules are hung on both sides of the {Ln(C2O4)1.5}n layer, forming a layer structure. As shown in Fig. 3a, the neighboring layers are further linked in the sequence of ⋯AAA⋯ through extensive hydrogen bonds including O2⋯O3b [2.592(5) Å], O4⋯O7c [2.678(5) Å], N1⋯O3d [2.770(6) Å] (symmetry codes: b = −x + 1, −y + 1, −z + 2; c = x, y − 1, z + 1; d = −x + 1, −y, −z + 2), thus forming a three dimensional supramolecular structure (Fig. 3a). The 8-hydroxyquinoline rings from neighboring layers are parallel displaced with respect to each other, and reveal aromatic stacking interactions with a displacement angle of 27.8(1)° and a plane⋯plane distance of 3.220(1) Å (Fig. 3b, Table S1†).19
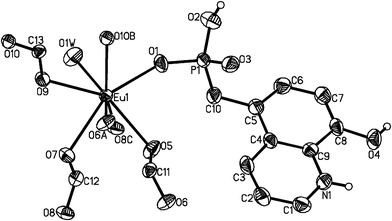 |
| | Fig. 1 The asymmetric unit of structure 1 with the atomic labeling scheme (50% probability). Lattice water O2W, O3W and all H atoms except attached to N1, O2 and O4 atoms are omitted for clarity. | |
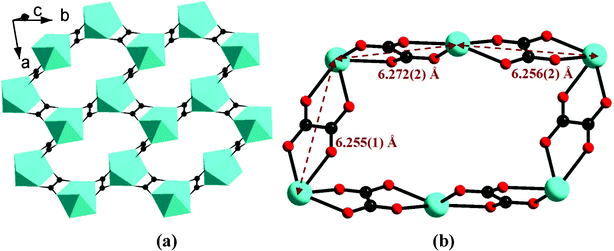 |
| | Fig. 2 The lanthanide oxalate layer of {Ln(C2O4)1.5}n (a) and the parallelogram-like 24-membered ring (b) in compound 1. | |
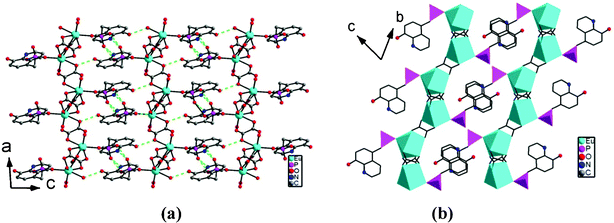 |
| | Fig. 3 The packing structures with H–bond interactions (a) and aromatic stacking interactions (b) in 1. | |
The structures of 2–4 are analogous to 1 except that the Eu(III) in 1 is replaced by Gd(III) in 2, Tb(III) in 3, and Dy(III) in 4, respectively. The crystal cell volume decreases in the sequence 1 > 2 > 3 > 4, in agreement with the decreasing sequence of ionic radii of the corresponding Ln(III) ions. The Ln(III) ions are bridged by oxalate units to form {Ln(C2O4)1.5}n layers, where the Ln⋯Ln distances across the oxalate unit are in the range of 6.227(1)–6.248(1) Å for 2, 6.188(1)–6.222(1) Å for 3, and 6.193(1)–6.224(1) Å for 4, respectively. The 8-hydroxyquinoline groups are arranged on two sides of the {Ln(C2O4)1.5}n layers due to the coordination of phosphonate oxygen (O1) to Ln(III) with Ln1–O1 distances of 2.235(3) Å in 2, 2.223(3) in 3, and 2.209(3) in 4, respectively. These layers are connected by hydrogen-bond interactions among phosphonate oxygen atoms O2 and O3, oxalate oxygen O7, hydroxyl oxygen atom O4, and pyridine nitrogen atom N1 (Table 3), and forming similar three dimensional supramolecular structure to that of 1. The aromatic stacking interactions between parallel 8-hydroxyquinoline rings exhibit a displacement angle of 28.0(1)° and a plane⋯plane distance of 3.213(1) Å in 2, 28.2(1)° and 3.202(1) Å in 3, and 27.8(1)° and 3.218(1) Å in 4, respectively (Table S1).
Table 3 Hydrogen bonds in 1–4 (Å and °)
|
Symmetry codes = x, y + 1, z.
= −x + 1, −y + 1, −z + 2.
= x, y − 1, z + 1.
= −x + 1, −y, −z + 2.
= −x + 2, −y + 1, −z + 1.
|
| |
1
|
2
|
| D–H⋯A |
d(D⋯A) |
< (DHA) |
d(D⋯A) |
< (DHA) |
| O1W–H1WA⋯O2Wa |
2.746(6) |
167.2(1) |
2.738(4) |
166.4(1) |
| O2–H2A⋯O3b |
2.592(5) |
165.2(1) |
2.590(5) |
164.7(1) |
| O4–H4A⋯O7c |
2.678(5) |
170.9(1) |
2.687(4) |
171.2(1) |
| N1–H1A⋯O3d |
2.770(6) |
152.6(1) |
2.771(5) |
152.5(1) |
| O2W–H2WD⋯O1We |
2.981(6) |
120.7(1) |
2.986(5) |
121.9(1) |
| |
3
|
4
|
| D–H⋯A |
d(D⋯A) |
< (DHA) |
d(D⋯A) |
< (DHA) |
| O1W–H1WA⋯O2Wa |
2.732(5) |
166.6(1) |
2.730(5) |
166.1(1) |
| O2–H2A⋯O3b |
2.583(5) |
164.3(1) |
2.598(5) |
163.8(1) |
| O4–H4A⋯O7c |
2.683(4) |
170.5(1) |
2.680(4) |
170.4(1) |
| N1–H1A⋯O3d |
2.791(5) |
152.9(1) |
2.787(5) |
153.8(1) |
| O2W–H2WD⋯O1We |
2.977(5) |
122.6(1) |
2.985(5) |
122.9(1) |
So far, most of the reported lanthanide oxalatophosphonates reveal three dimensional structures,3,18,20 and only few cases show layer structures including [Ln4(C2O4)5(2-pmpH)2(H2O)7]·5H2O [Ln = Gd, Tb, Dy; 2-pmpH2 = 2-pyridylmethylphosphonicacid],21 [Ln3(Hpmp)3(C2O4)2(H2O)5](ClO4)2·9H2O [Ln(III) = Gd, Eu, Tb; H2pmp = N-piperidinomethane-1-phosphonic acid],16 and [Ln(H3L)(C2O4)]·2H2O [Ln(III) = La–Dy, Er and Y, H4L = C6H5CH2N(CH2PO3H2)2].17 These compounds reveal clearly different layer structures from title compounds. In [Ln4(C2O4)5(2-pmpH)2(H2O)7]·5H2O, the double layer structures were observed, where net-like {Ln4(ox)5}n layers are connected by phosphonate oxygens. [Ln3(Hpmp)3(C2O4)2(H2O)5](ClO4)2·9H2O have a microporous layer, where each [Ln6(Hpmp)6] unit is linked to six neighboring equivalents by eight C2O42− ligands. In [Ln(H3L)(C2O4)]·2H2O, two {LnO8} polyhedra and four {PO3C} tetrahedra are interconnected into a subunit through corner-sharing, and these subunits are further bridged by the oxalate anions into a layer. Consequently, compounds 1–4 provide a kind of new layer structure in lanthanide oxalatophosphonate materials.
Magnetic and fluorescent properties
The temperature-dependent magnetic susceptibilities of 1–4 were studied in the temperature range of 1.8–300 K at the magnetic field of 1000 Oe. As shown in Fig. 4, the χMT value of 1 at 300 K (1.36 cm3 K mol−1) is much higher than the theoretical value of 0 cm3 K mol−1 for one isolated Eu(III) ion (S = 3, L = 3, 7F0), because not only the ground state (7F0) but also the first (7F1) and even higher exited states can be populated at room temperature.13 Upon lowering the temperature, the χMT value gradually decreases to 0.013 cm3 K mol−1 at 1.8 K, corresponding to a nonmagnetic ground state of 7F0 for Eu(III) ions. The similar magnetic behaviors were also observed in other Eu(III) compounds.22 For compound 2, the χMT value at 300 K is 8.04 cm3 K mol−1, close to the value of 7.88 cm3 K mol−1 for one isolated Gd(III) ion (S = 7/2). Upon cooling, the χMT value slowly decreases to 7.86 cm3 K mol−1 at 14 K, and then rapidly reaches 7.23 cm3 K mol−1 at 1.8 K. This feature indicates the presence of very weak antiferromagnetic interactions between Gd(III) ions, which is further confirmed by a small Weiss constant θ = −0.36 K (Fig. S3†). For 3 and 4, the observed χMT values at 300 K are 12.09, and 14.60 cm3 K mol−1, respectively, which are comparable to 11.82 cm3 K mol−1 for one isolated Tb(III) ion (J = 6, g = 3/2), and 14.17 cm3 K mol−1 for one uncoupled Dy(III) ion (J = 15/2, g = 4/3), respectively.23 Upon cooling, the χMT values of both 3 and 4 remain nearly constants before 60 K, and then dramatically decrease to 4.45 and 8.74 cm3 K mol−1 at 1.8 K, respectively. The decrease in χMT values with lowering the temperature can be attributed to the depopulation of the Stark levels together with weak antiferromagnetic interactions between the lanthanide ions.23,24 In the whole temperature range, the magnetic data were fitted according to the Curie–Weiss law, giving Weiss constant −3.40 K for 3 and −1.41 K for 4, respectively (Fig. S4 and S5†).
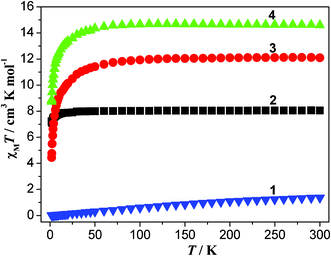 |
| | Fig. 4 The χMT vs. T plots for 1–4. | |
The solid-state fluorescent properties of 1–4 and 5pm8hqH3·HCl·H2O were investigated at room temperature under the excitation at 409 nm for 1, 360 nm for 2 and 5pm8hqH3·HCl·H2O, 390 nm for 3, and 387 nm for 4. As shown in Fig. 5, compounds 1–4 show the ligand-centered (LC) fluorescence with emission band at 529 nm for 1, 524 nm for 2, 523 nm for 3, and 523 nm for 4, confirmed by the broad emission band of 5pm8hqH3·HCl·H2O (λmax = 523 nm).25 In contrast to 5pm8hqH3·HCl·H2O, 1–4 reveal enhanced fluorescence, which might be due to the aromatic stacking interactions between 8-hydroxyquinoline rings.26 Exception the ligand-centered (LC) emission, the weak Ln(III)-centred emissions were found in 1 and 4, showing the characteristic band of 614 nm attributed to 5D0 → 7F2 for Eu(III), and 580 nm attributed to 4D9/2 → 6H13/2 for Dy(III), respectively.2c,17 The fluorescent lifetimes of 1, 3 and 4 were measured. However, only the lifetimes from the ligand-centered (LC) fluorescence were obtained, 1.5 ns for 1, 1.8 ns for 3, and 1.6 ns for 4.
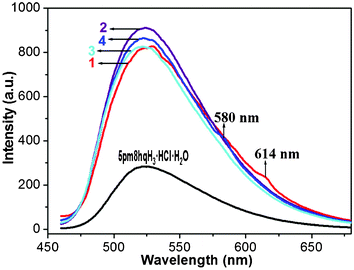 |
| | Fig. 5 Solid-state emission spectra for compounds 1–4 and 5pm8hqH3·HCl·H2O at room temperature. | |
Conclusions
5-Phosphonomethyl-8-hydroxyquinoline (5pm8hqH3) and its lanthanide oxalatophosphonates Ln(5pm8hqH3)(C2O4)1.5(H2O)·2H2O [Ln(III) = Eu(1), Gd(2), Tb(3), Dy(4)] have been synthesized. Compounds 1–4 show isostructural layer structures, where 5pm8hqH3 ligands are hung on both sides of the {Ln(C2O4)1.5}n layer through the coordination of one phosphonate oxygen to the Ln(III) ion. Neighboring layers are linked through hydrogen bonds and aromatic stacking of the 8-hydroxyquinoline rings into a supramolecular 3D structure. Magnetic studies indicate the presence of weak antiferromagnetic interactions in 2, and the coexistence of depopulation of the Stark levels and weak antiferromagnetic interactions in 1, 3 and 4. In solid-state fluorescent measurements, 1–4 show stronger ligand-centered emissions than that of 5pm8hqH3·HCl·H2O. In addition, 1 and 4 reveal the Ln(III)-centered emissions although their intensities are weak.
Acknowledgements
We thank financial support from the NSF of China (No. 90922006), the NSF of Jiangsu Province (No. BK2009009), and the NSF of China (No. 21171090).
References
-
(a) Q. Yue, J. Yang, G.-H. Li, G.-D. Li and J.-S. Chen, Inorg. Chem., 2006, 45, 4431 CrossRef CAS;
(b) J.-G. Mao, Coord. Chem. Rev., 2007, 251, 1493 CrossRef CAS;
(c) S.-S. Bao, L.-F. Ma, Y. Wang, L. Fang, C.-J. Zhu, Y.-Z. Li and L.-M. Zheng, Chem.–Eur. J., 2007, 13, 2333 CrossRef CAS;
(d) L. Cunha-Silva, S. Lima, D. Ananias, P. Silva, L. Mafra, L. D. Carlos, M. Pillinger, A. A. Valente, F. A. Almeida Paz and J. Rocha, J. Mater. Chem., 2009, 19, 2618 RSC;
(e) Z. Amghouz, S. Garcia-Granda, J. R. Garcia, A. Clearfield and R. Valiente, Cryst. Growth Des., 2011, 11, 5289 CrossRef CAS.
-
(a) Z.-Y. Du, H.-B. Xu and J.-G. Mao, Inorg. Chem., 2006, 45, 9780 CrossRef CAS;
(b) X. Li, Q. Liu, J. Lin, Y. Li and R. Cao, Inorg. Chem. Commun., 2009, 12, 502 CrossRef CAS;
(c) T.-H. Zhou, F.-Y. Yi, P.-X. Li and J.-G. Mao, Inorg. Chem., 2010, 49, 905 CrossRef CAS.
- S.-M. Ying and J.-G. Mao, Cryst. Growth Des., 2006, 964 CAS.
-
(a) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS , and some references therein;
(b) L. Z. Zhang, W. Gu, B. Li, X. Liu and D. Z. Liao, Inorg. Chem., 2007, 46, 622 CrossRef CAS.
- H. L. Ngo and W.-B. Lin, J. Am. Chem. Soc., 2002, 124, 14298 CrossRef CAS.
-
(a) D.-K. Cao, S.-Z. Hou, Y.-Z. Li and L.-M. Zheng, Cryst. Growth Des., 2009, 9, 4445 CrossRef CAS;
(b) J.-L. Song, F.-Y. Yi and J.-G. Mao, Cryst. Growth Des., 2009, 9, 3273 CrossRef CAS.
-
(a) X.-G. Liu, K. Zhou, J. Dong, C.-J. Zhu, S.-S. Bao and L.-M. Zheng, Inorg. Chem., 2009, 48, 1901 CrossRef CAS;
(b) S.-F. Tang, J.-L. Song, X.-L. Li and J.-G. Mao, Cryst. Growth Des., 2007, 7, 360 CrossRef CAS.
-
(a) D.-K. Cao, Y.-Z. Li, Y. Song and L.-M. Zheng, Inorg. Chem., 2005, 44, 3599 CrossRef CAS;
(b) J. Galezowska, R. Janicki, H. Kozlowski, A. Mondry, P. Mlynarz and L. Szyrwiel, Eur. J. Inorg. Chem., 2010, 1696 CrossRef CAS.
-
(a) M. Albrecht, R. Frohlich, J.-C. G. Bunzli, A. Aebischer, F. Gumy and J. Hamacek, J. Am. Chem. Soc., 2007, 129, 14178 CrossRef CAS;
(b) M. Albrecht, M. Fiege and O. Osetska, Coord. Chem. Rev., 2008, 252, 812 CrossRef CAS;
(c) M. Albrecht, O. Osetska, J.-C. G. Bunzli, F. Gumy and R. Frohlich, Chem.–Eur. J., 2009, 15, 8791 CrossRef CAS;
(d) G. Bozoklu, C. Marchal, J. Pecaut, D. Imbert and M. Mazzanti, Dalton Trans., 2010, 39, 9112 RSC.
-
(a) H.-B. Xu, J. Li, L.-X. Shia and Z.-N. Chen, Dalton Trans., 2011, 40, 5549 RSC;
(b) H.-B. Xu, H.-M. Wen, Z.-H. Chen, J. Li, L.-X. Shi and Z.-N. Chen, Dalton Trans., 2010, 39, 1948 RSC;
(c) H.-B. Xu, X.-M. Chen, Q.-S. Zhang, L.-Y. Zhang and Zh.-N. Chen, Chem. Commun., 2009, 7318 RSC.
- S. P. Man, D. M. Benoit, E. Buchaca, F. Esan, M. Motevalli, J. Wilson and A. Sullivan, Inorg. Chem. Commun., 2006, 45, 5328 CAS.
- J. H. Burckhalter and R. I. Leib, J. Org. Chem., 1961, 26, 4078 CrossRef CAS.
-
O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
-
SAINT, Program for Data Extraction and Reduction, Siemens Analytical X-ray Instruments, Madison, WI, 1994–1996 Search PubMed.
-
(a)
SHELXTL, Reference Manual, version 5.0, Siemens Industrial Automation, Analytical Instruments, Madison, WI, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
- X. Li, T. Liu, Q. Lin and R. Cao, Cryst. Growth Des., 2010, 10, 608 CAS.
- Y.-Y. Zhu, Z.-G. Sun, F. Tong, Z.-M. Liu, C.-Y. Huang, W.-N. Wang, C.-Q. Jiao, C.-L. Wang, C. Li and K. Chen, Dalton Trans., 2011, 40, 5584 RSC.
- L. Liu, Z.-G. Sun, N. Zhang, Y.-Y. Zhu, Y. Zhao, X. Lu, F. Tong, W.-N. Wang and C.-Y. Huang, Cryst. Growth Des., 2010, 10, 406 CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
-
(a) J.-L. Song and J.-G. Mao, Chem.–Eur. J., 2005, 11, 1417 CrossRef CAS;
(b) Y.-L. Huang, M.-Y. Huang, T.-H. Chan, B.-C. Chang and K.-H. Lii, Chem. Mater., 2007, 19, 3232 CrossRef CAS;
(c) Y. Zhao, J. Li, Z.-G. Sun, J. Zhang, Y.-Y. Zhu, X. Lu, L. Liu and N. Zhang, Inorg. Chem. Commun., 2008, 11, 1057 CrossRef CAS;
(d) Y.-Y. Zhu, Z.-G. Sun, Y. Zhao, J. Zhang, X. Lu, N. Zhang, L. Liu and F. Tong, New J. Chem., 2009, 33, 119 RSC;
(e) Y.-Y. Zhu, Z.-G. Sun, H. Chen, J. Zhang, Y. Zhao, N. Zhang, L. Liu, X. Lu, W.-N. Wang, F. Tong and L.-C. Zhang, Cryst. Growth Des., 2009, 9, 3228 CrossRef CAS;
(f) L. Zhang, S. Xu, Y. Zhou, X. Zheng, C. Yu, Z. Shi, S. Hassan and C. Chen, CrystEngComm, 2011, 13, 6511 RSC.
- T.-H. Yang, D.-K. Cao, Y.-Z. Li and L.-M. Zheng, J. Solid State Chem., 2010, 183, 1159 CrossRef CAS.
-
(a) Q.-Y. Liu, W.-F. Wang, Y.-L. Wang, Z.-M. Shan, M.-S. Wang and J. Tang, Inorg. Chem., 2012, 51, 2381 CrossRef CAS;
(b) X.-Y. Qian, J.-H. Zhang, T.-H. Zhoua and J.-G. Mao, Dalton Trans., 2012, 41, 1229 RSC;
(c) M.-F. Wu, M.-S. Wang, S.-P. Guo, F.-K. Zheng, H.-F. Chen, X.-M. Jiang, G.-N. Liu, G.-C. Guo and J.-S. Huang, Cryst. Growth Des., 2011, 11, 372 CrossRef CAS.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS.
-
(a)
J.-C. G. Bunzli and G. R. Chopin, Lanthanide Probes in Life, Chemical and Earth Sciences, Elsevier, Amsterdam, 1989 Search PubMed;
(b) J.-P. Costes and F. Nicodeme, Chem.–Eur. J., 2002, 8, 3442 CrossRef CAS.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- S.-J. Yoon, J. H. Kim, J. W. Chung and S. Y. Park, J. Mater. Chem., 2011, 21, 18971 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The yield, elemental analyses, and IR data for 1–4, the distances and angles for aromatic stacking interactions in 1–4, X-ray crystallographic files in CIF format, XRD patterns, TG curves, and the χM−1vs. T curves for 2–4. CCDC reference numbers 810422–810425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20111a |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.