DOI:
10.1039/C2RA20105D
(Paper)
RSC Adv., 2012,
2, 5254-5264
Mononuclear palladium and heterodinuclear palladium–ruthenium complexes of semicarbazone ligands. Synthesis, characterization, and application in C–C cross-coupling reactions†
Received
18th January 2012
, Accepted 23rd March 2012
First published on 23rd March 2012
Abstract
Reaction of salicylaldehyde semicarbazone (L1), 2-hydroxyacetophenone semicarbazone (L2), and 2-hydroxynaphthaldehyde semicarbazone (L3) with [Pd(PPh3)2Cl2] in ethanol in the presence of a base (NEt3) affords a family of yellow complexes (1a, 1b and 1c, respectively). In these complexes the semicarbazone ligands are coordinated to palladium in a rather unusual tridentate ONN-mode, and a PPh3 also remains coordinated to the metal center. Crystal structures of the 1b and 1c complexes have been determined, and structure of 1a has been optimized by a DFT method. In these complexes two potential donor sites of the coordinated semicarbazone, viz. the hydrazinic nitrogen and carbonylic oxygen, remain unutilized. Further reaction of these palladium complexes (1a, 1b and 1c) with [Ru(PPh3)2(CO)2Cl2] yields a family of orange complexes (2a, 2b and 2c, respectively). In these heterodinuclear (Pd–Ru) complexes, the hydrazinic nitrogen (via dissociation of the N–H proton) and the carbonylic oxygen from the palladium-containing fragment bind to the ruthenium center by displacing a chloride and a carbonyl. Crystal structures of 2a and 2c have been determined, and the structure of 2b has been optimized by a DFT method. All the complexes show characteristic 1H NMR spectra and, intense absorptions in the visible and ultraviolet region. Cyclic voltammetry on all the complexes shows an irreversible oxidation of the coordinated semicarbazone within 0.86–0.93 V vs. SCE, and an irreversible reduction of the same ligand within −0.96 to −1.14 V vs. SCE. Both the mononuclear (1a, 1b and 1c) and heterodinuclear (2a, 2b and 2c) complexes are found to efficiently catalyze Suzuki, Heck and Sonogashira type C–C coupling reactions utilizing a variety of aryl bromides and aryl chlorides. The Pd–Ru complexes (2a, 2b and 2c) are found to be better catalysts than the Pd complexes (1a, 1b and 1c) for Suzuki and Heck coupling reactions.
Introduction
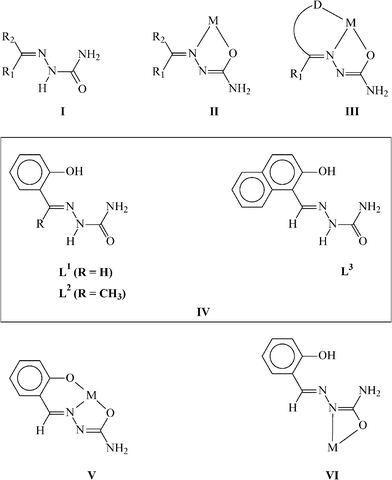
The chemistry of transition metal complexes of the semicarbazone ligands has been receiving significant attention, mainly due to their bioinorganic relevance.1 A large majority of the semicarbazone complexes have found wide medicinal applications because of their potentially beneficial biological (viz. antibacterial, antimalarial, antiviral, and antitumor) activities.2 Studies on the binding of semicarbazones to different transition metal ions are of considerable importance in this respect. However, we were attracted to the chemistry of semicarbazone complexes of the platinum metals primarily because of the variable coordination modes displayed by these ligands in their complexes.3 Semicarbazones (I, Chart 1) have been observed to bind to a metal ion as bidentate NO-donors, forming five-membered chelate rings (II).3b,3c If a third donor site (D) is incorporated into such ligands, linked to the carbonylic carbon via one or two intervening atoms, then DNO-tricoordination (III) normally takes place.4 For the present work, we have chosen three potentially tridentate semicarbazones (IV), viz. the semicarbazone of salicylaldehyde, 2-hydroxyacetophenone and 2-hydroxynaphthaldehyde. Individual ligand abbreviations are shown in (IV). To interact with the chosen semicarbazones palladium has been selected as the metal center, mainly because of the world-wide fame of its complexes for serving as precatalysts in bringing about C–C coupling reactions of different types.5 The efficiency of different palladium complexes as precatalysts is largely dictated by the coordination environment around the metal center, and hence we were interested to see how binding by the chosen semicarbazone ligands (IV) can influence the catalytic property of the palladium center. It may be mentioned here that, although the chemistry of some semicarbazones with different metals has been studied,6 that of the chosen semicarbazones (IV) with palladium appears to have remained unexplored. The salicylaldehyde semicarbazone (as well as the other two ligands) is known to coordinate a metal center, via dissociation of the hydrazinic NH and phenolic OH protons, as a dianionic ONO-donor (V).4 It has also been observed to coordinate as a bidentate NO-donor, via dissociation of only the hydrazinic proton, forming a rather uncommon four-membered chelate ring (VI).3c With an expectation of inducing a ONO-mode of binding of the semicarbazones (as in V), their reactions have been carried out with [Pd(PPh3)2Cl2]. This particular complex has been selected as the palladium starting material because of its demonstrated ability to incorporate dianionic tridentate ligands via elimination of two chlorides and a triphenylphosphine,7 and the expected presence of the remaining triphenylphosphine was also expected to facilitate catalytic activity of the resulting complex. The reactions have indeed afforded a family of three mixed-ligand palladium complexes with the expected composition, but the semicarbazones have displayed an unexpected mode of coordination. Due to this unusual binding mode of the semicarbazones, these palladium complexes could be utilized as efficient building blocks for the synthesis of a group of interesting heterodinuclear complexes incorporating ruthenium as the second metal center. The catalytic activity towards different C–C coupling reactions of both the Pd and Pd–Ru complexes has also been investigated and compared. Herein we report the chemistry of both the mononuclear and heterodinuclear complexes, with special reference to their formation, structure and catalytic activity for C–C coupling reactions using a variety of aryl bromides and chlorides.
 |
| | Chart 1 | |
Results and discussion
Syntheses and structures
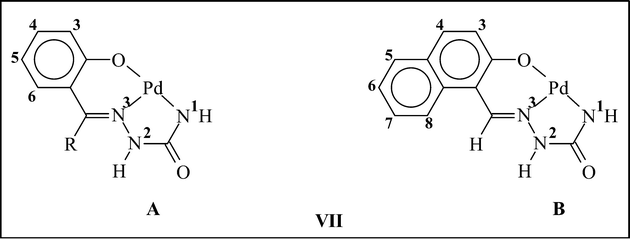
As outlined in the introduction, a major interest of the present study has been to examine how the selected semicarbazones (IV) react with the metal center in [Pd(PPh3)2Cl2] and bind to it. Reactions of the chosen semicarbazones (L1, L2 and L3) with [Pd(PPh3)2Cl2] have afforded a family of yellow complexes (1a, 1b and 1c, respectively) in decent yields. Preliminary (microanalytical and spectroscopic) characterizations of these complexes, although they indicated the presence of a semicarbazone and a triphenylphosphine in the coordination sphere of palladium, failed to predict any definite binding mode of the semicarbazone in them. For an unambiguous characterization of these complexes, structure of a selected member of the family, viz.1b, has been determined by X-ray crystallography. The structure is shown in Fig. 1 and selected bond parameters are listed in Table 1. The structure reveals that the semicarbazone is coordinated to palladium in a rather unexpected dianionic tridentate ONN-fashion (VIIA; R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3), via loss of the phenolic proton and one NH2 proton, forming two adjacent six- and five-membered chelate rings with O–Pd–N and N–Pd–N bite angles of 92.60(11)° and 80.67(12)°, respectively. A triphenylphosphine is also coordinated to the metal center, which is trans to the central imine-nitrogen of the coordinated semicarbazone. Palladium is thus nested in an ON2P core, which is slightly distorted from ideal square-planar geometry, as reflected in the bond parameters around the metal center. The Pd–O, Pd–N and Pd–P distances are all quite normal,4 and so are the bond distances within the coordinated semicarbazone.3c
CH3), via loss of the phenolic proton and one NH2 proton, forming two adjacent six- and five-membered chelate rings with O–Pd–N and N–Pd–N bite angles of 92.60(11)° and 80.67(12)°, respectively. A triphenylphosphine is also coordinated to the metal center, which is trans to the central imine-nitrogen of the coordinated semicarbazone. Palladium is thus nested in an ON2P core, which is slightly distorted from ideal square-planar geometry, as reflected in the bond parameters around the metal center. The Pd–O, Pd–N and Pd–P distances are all quite normal,4 and so are the bond distances within the coordinated semicarbazone.3c
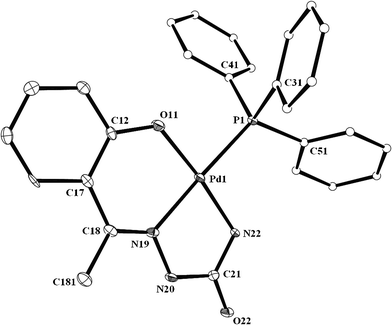 |
| | Fig. 1 A view of the 1b complex. (One of the two independent molecules is shown) | |
Table 1 Selected bond distances and bond angles for 1b and 2a
|
1b
|
| Bond distances (Å) |
| Pd–P |
2.2686(9) |
C(12)–O(11) |
1.312(4) |
| Pd–O(11) |
1.984(2) |
C(18)–N(19) |
1.292(4) |
| Pd–N(19) |
2.033(3) |
N(19)–N(20) |
1.386(4) |
| Pd–N(22) |
1.976(3) |
N(20)–C(21) |
1.378(4) |
| |
|
C(21)–N(22) |
1.336(5) |
| |
|
C(21)–O(22) |
1.245(4) |
| Bond angles (°) |
| N(19)–Pd–P |
176.63(8) |
O(11)–Pd–N(19) |
92.60(11) |
| O(11)–Pd–N(22) |
172.97(12) |
N(19)–Pd–N(22) |
80.67(12) |
| |
|
2a
|
| Bond distances (Å) |
| Pd–P(1) |
2.2650(5) |
C(1)–O(1) |
1.312(3) |
| Pd–O(1) |
1.9984(17) |
C(7)–N(1) |
1.287(3) |
| Pd–N(1) |
2.0171(16) |
N(1)–N(2) |
1.360(2) |
| Pd–N(3) |
1.9850(19) |
N(2)–C(8) |
1.353(3) |
| Ru–P(2) |
2.3793(5) |
C(8)–N(3) |
1.324(3) |
| Ru–P(3) |
2.3843(5) |
C(8)–O(2) |
1.295(3) |
| Ru–Cl |
2.3992(5) |
C(9)–O(3) |
1.148(3) |
| Ru–C(9) |
1.824(2) |
|
|
| Ru–N(2) |
2.0651(16) |
|
|
| Ru–O(2) |
2.2424(15) |
|
|
| Bond angles (°) |
| N(1)–Pd–P(1) |
176.58(5) |
N(2)–Ru–Cl |
161.24(5) |
| O(1)–Pd–N(3) |
172.93(8) |
O(2)–Ru–C(9) |
165.42(7) |
| N(1)–Pd–O(1) |
93.10(7) |
P(2)–Ru–P(3) |
176.83(2) |
| N(1)–Pd–N(3) |
80.50(7) |
O(2)–Ru–N(2) |
60.97(6) |
Participation of the terminal NH2 fragment of the semicarbazone in coordination to palladium via loss of one proton, as observed in the structurally characterized 1b complex, is rather unusual and, to our knowledge, unprecedented. To ensure that the same ONN-mode of binding also exists in the other two complexes, the structure of another member of this family, viz.1c, has also been determined by X-ray crystallography. The structure (Fig. S1, ESI†) clearly shows that the semicarbazone ligand is coordinated to palladium in the similar ONN-mode (VIIB) as in the 1b complex, and all the structural features of 1c (Table S1, ESI†) are found to be similar to those observed for 1b. Though the crystal structure of 1a has not been solved, its structure has been optimized by a DFT method.8,9 The optimized structure of 1a and the evaluated bond parameters (Fig. S2 and Table S2, ESI†) are found to be very similar to those of 1b.
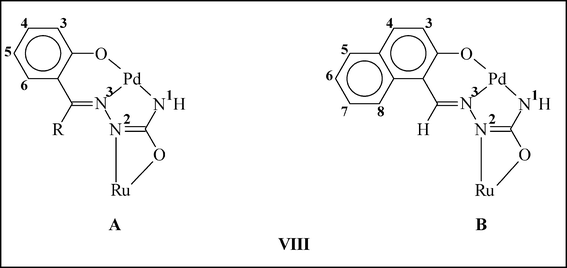
The three palladium complexes (1a, 1b and 1c) represent a unique family of semicarbazone complexes where the hydrazinic N–H proton did not undergo dissociation and the carbonylic-oxygen did not participate in coordination, instead, proton dissociation and coordination took place from the NH2 fragment. Availability of the two unutilized donor sites of the coordinated semicarbazone, viz. the hydrazinic nitrogen and carbonylic oxygen, makes these palladium complexes potential building blocks for the synthesis of multinuclear complexes. This possibility has been explored by carrying out further reaction of these palladium complexes with [Ru(PPh3)2(CO)2Cl2]. This particular ruthenium complex has been chosen because of its demonstrated ability to undergo facile reaction with the –N(H)–C(O)– fragment of the semicarbazone ligand.3c The reactions have afforded a family of orange complexes (2a, 2b and 2c, respectively) in decent yields. The structure of one selected member of the family, viz.2a, has been determined by X-ray crystallography. The structure (Fig. 2) shows that the semicarbazone ligand, which was already coordinated to palladium in the ONN-fashion, is also linked to the ruthenium center through the hydrazinic-nitrogen (via dissociation of the proton) and the carbonylic-oxygen, forming a four-membered chelate ring (VIIIA; RH) with N–Ru–O bite angle of 60.97(6)°. Compared to the mononuclear Pd complex, the C–O and C–N distances in the four-membered chelate ring of the Pd–Ru complex (Table 1) are found to be significantly different, which is attributable to binding to ruthenium via dissociation of the hydrazinic proton and the possible resonance (–N–C(O)– ↔ –NC(–O)–) in this fragment. Two triphenylphosphines, a carbonyl and a chloride remain coordinated to the ruthenium center. Ruthenium thus has a CNOP2Cl coordination sphere, which is distorted octahedral in nature. The two PPh3 ligands are mutually trans. The carbonyl is trans to the oxygen and the chloride is trans to the nitrogen. To ensure that similar bridging mode of binding of the semicarbazones exists also in 2b and 2c, crystal structure of another member of this family, viz.2c, has also been determined. The structure (Fig. S3, ESI†) clearly shows the same type of binding mode (VIIIB) as in 2a, and observed bond parameters (Table S1, ESI†) also compare well with those found in 2a. While crystal structure of 2b has not been determined, its structure has been optimized by DFT method. The optimized structure of 2b and the evaluated bond parameters (Fig. S4 and Table S2, ESI†) are found to be very similar to those of 2a.
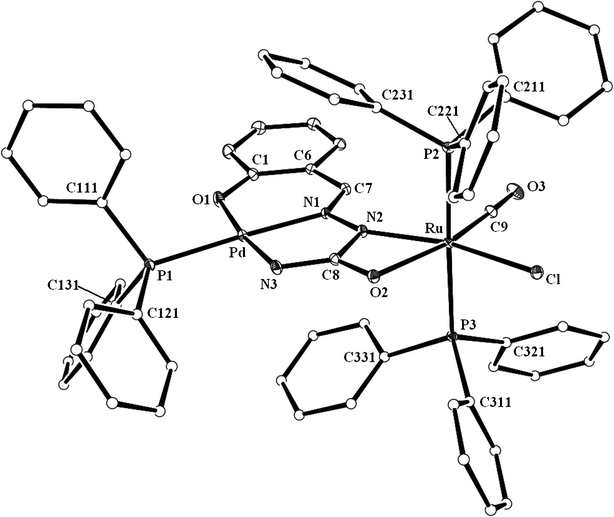 |
| | Fig. 2 A view of the 2a complex. | |
Spectral and electrochemical properties
All the palladium (1a, 1b and 1c) and Pd–Ru (2a, 2b and 2c) complexes are diamagnetic, which corresponds to the bivalent states of palladium (low-spin d8, S = 0) and ruthenium (low-spin d6, S = 0) in them. 1H NMR spectra of these complexes have been recorded in CDCl3 solution. In the spectra of the palladium complexes, signals for the coordinated triphenylphosphines are observed as broad peaks within 7.3–7.8 ppm. In complexes 1a and 1c the azomethine proton signal of the coordinated semicarbazone is observed at 7.72 and 7.84 ppm, respectively. The methyl signal from the coordinated semicarbazone in the 1b complex is observed at 2.32 ppm. Signals for the hydrazinic NH and the palladium-bound NH of the semicarbazone are observed at around 8.0 ppm and 11.0 ppm, respectively. Most of the expected signals from the aryl fragment of the coordinated semicarbazone are clearly observed in the aromatic region, while few could not be detected due to their overlap with other signals. Besides disappearance of the hydrazinic NH signal, the 1H NMR spectrum of the each Pd–Ru complex is found to be qualitatively similar to that of its palladium precursor complex. The 1H NMR spectral data of the palladium (1a, 1b and 1c) and Pd–Ru (2a, 2b and 2c) complexes are therefore consistent with their composition.
Infrared spectra of the palladium (1a, 1b and 1c) complexes show many bands of varying intensities within 4000–400 cm−1. Assignment of each individual band to a specific vibration has not been attempted. However, three strong bands, displayed near 536, 692 and 744 cm−1, are attributed to the coordinated PPh3 ligands. Comparison with the spectrum of [Pd(PPh3)2Cl2] shows the presence of several new bands (e.g. near 820, 1330, 1400, 1540, 1630, 3053 and 3400 cm−1) in the spectra of the 1a, 1b and 1c complexes, which must be due to the coordinated semicarbazone ligand. The infrared spectral features of the Pd–Ru (2a, 2b and 2c) complexes are mostly similar to those observed in their precursor (1a, 1b and 1c) complexes. An intense band observed near 1920 cm−1 in the Pd–Ru complexes is attributable to the ruthenium-bound carbonyl. The sharp band near 3400 cm−1, observed in all the three Pd complexes, is found to be absent in the Pd–Ru complexes, indicating loss of the hydrazinic N–H proton upon binding to ruthenium. The other sharp band, observed near 3050 cm−1 in all the three Pd complexes, is also found to be present in the Pd–Ru complexes, and thus this band may be attributed to the palladium-bound N–H fragment.
Electronic spectra of all the complexes have been recorded in dichloromethane solution. Spectral data are presented in Table 2. The palladium (1a, 1b and 1c) complexes show several intense absorptions in the visible and ultraviolet region. While the absorptions in the ultraviolet region are believed to be due to transitions within the ligand orbitals, origin of those in the visible region was unclear. To have an insight into the nature of absorptions in the visible region, DFT calculations have been performed on these complexes. The results are found to be qualitatively similar for all the three members of this group. Partial MO diagram for a representative palladium complex is shown in Fig. 3 and composition of selected molecular orbitals is given in Table 3. For the Pd complexes (1a, 1b and 1c) both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are delocalized almost entirely on the semicarbazone ligand. Hence the lowest energy absorption in the visible region is also assignable to a transition from a filled orbital to a vacant orbital of the semicarbazone ligand. DFT calculations on the Pd–Ru complexes (2a, 2b and 2c) again show (Table 3, Fig. 4) that the HOMO and LUMO have major contributions from the coordinated semicarbazone.10 Therefore, as observed in the Pd complexes, the lowest energy absorption in these Pd–Ru complexes is also due to a transition taking place within orbitals of the semicarbazone ligand.
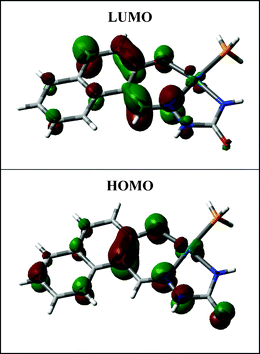 |
| | Fig. 3 A contour plot of complex 1c. | |
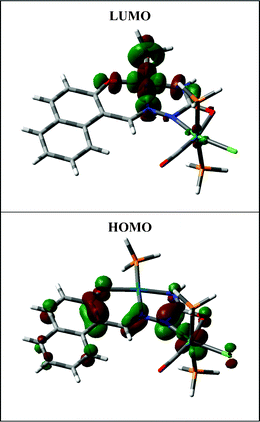 |
| | Fig. 4 A contour plot of complex 2c. | |
Table 2 Electronic spectral and cyclic voltammetric data of the complexes
| Compound |
Electronic spectral dataaλmax, nm (ε, M−1cm−1) |
Cyclic voltammetric databE, V |
In dichloromethane solution.
Solvent: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 dichloromethane-acetonitrile; supporting electrolyte: TBHP; reference electrode, SCE; scan rate: 50 mVs−1.
Shoulder.
Anodic peak potential (Epa).
Cathodic peak potential (Epc). :9 dichloromethane-acetonitrile; supporting electrolyte: TBHP; reference electrode, SCE; scan rate: 50 mVs−1.
Shoulder.
Anodic peak potential (Epa).
Cathodic peak potential (Epc).
|
|
1a
|
377c (2100), 291c (5100), 242c (14 900) |
0.86,d −1.14e |
|
1b
|
373c (2100), 302c (5100), 250c (14 500) |
0.93,d −1.12e |
|
1c
|
415c (5000), 394 (5300), 325c (12 900), 257c (35 300) |
0.89,d −1.14e |
|
2a
|
396 (5600), 297c (8700), 254c (22 400) |
0.87,d −1.08e |
|
2b
|
402 (5900), 305c (9200), 256c (22 600) |
0.90,d −0.96e |
|
2c
|
437c (15 500), 414c (15 200), 316c (18 600), 253c (55 600) |
0.88,d −0.97e |
Table 3 Composition of selected molecular orbitals
| Compound |
Contributing fragmentsc |
% contribution of fragments to |
| HOMO |
LUMO |
|
PPh3 bound to palladium center.
PPh3 bound to ruthenium center.
L1, L2 and L3 mean semicarbazones bound to metal center(s).
|
|
1a
|
Pd |
12.00 |
1.30 |
| L1 |
87.78 |
98.30 |
| PPh3a |
0.22 |
0.40 |
|
1b
|
Pd |
5.39 |
2.30 |
| L2 |
94.30 |
97.10 |
| PPh3a |
0.31 |
0.60 |
|
1c
|
Pd |
4.50 |
1.10 |
| L3 |
94.52 |
98.73 |
| PPh3a |
0.98 |
0.17 |
|
2a
|
Pd |
1.35 |
18.94 |
| Ru |
15.40 |
5.44 |
| L1 |
79.19 |
60.82 |
| Cl |
3.52 |
0.22 |
| PPh3a |
0.06 |
13.93 |
| PPh3b |
0.48 |
0.65 |
|
2b
|
Pd |
1.04 |
20.45 |
| Ru |
13.21 |
5.23 |
| L2 |
82.43 |
61.54 |
| Cl |
2.82 |
0.34 |
| PPh3a |
0.23 |
12.13 |
| PPh3b |
0.27 |
0.31 |
|
2c
|
Pd |
0.29 |
16.67 |
| Ru |
7.11 |
5.45 |
| L3 |
91.00 |
67.91 |
| Cl |
1.34 |
0.21 |
| PPh3a |
0.05 |
9.30 |
| PPh3b |
0.21 |
0.46 |
To assess the redox properties of the ruthenium center in the Pd–Ru complexes (2a, 2b and 2c) cyclic voltammetry has been performed on them in 1:9 dichloromethane–acetonitrile solution (0.1 M TBHP).11 For a proper comparison, cyclic voltammetry has also been performed on the Pd complexes (1a, 1b and 1c). Voltammetric data are presented in Table 2. All the complexes show an irreversible oxidative response on the positive side of the SCE and an irreversible reductive one on the negative side. In view of the composition of the HOMO the oxidative response is assigned to oxidation of the coordinated semicarbazone. Similarly, in view of the composition of the LUMO the reductive response is assigned to reduction of the coordinated semicarbazone. Cyclic voltammetric studies thus show that the higher oxidation state of the ruthenium center in the Pd–Ru complexes is not accessible within the permissible voltage window.
Catalytic activity
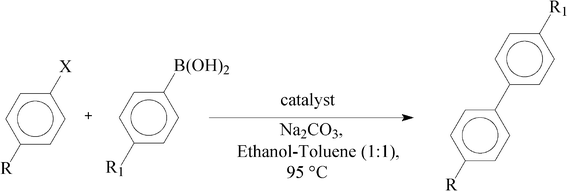
As described in the introduction, the primary objective of the present study has been to explore catalytic properties of the palladium complexes towards C–C coupling reactions. Hence the mononuclear palladium complexes (1a, 1b and 1c) were first tested as catalysts in the Suzuki coupling reactions between arylbromides and phenylboronic acids to yield the biphenyl products (Table 4). All the three complexes were found to successfully catalyze the coupling reactions, and their catalytic efficiency was also comparable.12 For example, using 1 mol% of 1a or 1b or 1c as the catalyst the Suzuki coupling of p-bromoacetophenone and phenylboronic acid was achieved, under relatively mild experimental conditions, in high yield (entries 1, 2 and 3). When the amount of catalyst was reduced to half, the reaction time needed to be doubled to get similar yield (entry 4). With electron-withdrawing substituents (e.g. CHO, CN) in the arylbromides, longer times (relative to COCH3 substituent) were required to achieve similar yields (entries 5 and 6). When p-cyanophenylboronic acid was used instead of phenylboronic acid, the coupling reactions (using 1c as catalyst) required a longer time and, at the same time, a sharp decrease in the yield was also noted (entries 7, 8 and 9). Encouraged by the facile C–C coupling of aryl bromides with phenylboronic acid, catalyzed by the palladium complexes, a similar coupling reaction via activation of C–Cl bond of aryl chlorides was also attempted. These mononuclear complexes (1a, 1b and 1c) were indeed found to bring about C–Cl bond activation of aryl chlorides and thus promote their coupling with phenylboronic acid (entries 10, 11 and 12). However, compared to the corresponding aryl bromides, a much longer time was needed for these reactions. A significant reduction in yield was observed upon changing the substituent from COCH3 to CHO to CN in the arylchlorides.
Table 4 Suzuki cross-coupling of aryl halides with 4-R1-phenylboronic acida
| Entry |
R |
R1 |
X |
Catalyst |
Time, h |
Amt of cat., mol (%) |
Conversion,b % |
|
Reaction conditions: aryl halide (1.0 mmol), 4-R1-phenylboronic acid (1.2 mmol), Na2CO3 (2.4 mmol), catalyst, ethanol–toluene (1:1) mixture (4 mL), 95 °C.
Determined by 1H NMR on the basis of residual aryl halide.20
1 mol% of both [Ru] and 1c catalysts were used.
|
| 1 |
COCH3 |
H |
Br |
1a
|
5.5 |
1 |
>99 |
| 2 |
COCH3 |
H |
Br |
1b
|
5.5 |
1 |
>99 |
| 3 |
COCH3 |
H |
Br |
1c
|
5.5 |
1 |
>99 |
| 4 |
COCH3 |
H |
Br |
1c
|
11 |
0.5 |
>99 |
| 5 |
CHO |
H |
Br |
1c
|
11 |
1 |
>99 |
| 6 |
CN |
H |
Br |
1c
|
12 |
1 |
>99 |
| 7 |
COCH3 |
CN |
Br |
1c
|
24 |
1 |
>99 |
| 8 |
CHO |
CN |
Br |
1c
|
24 |
1 |
37 |
| 9 |
CN |
CN |
Br |
1c
|
24 |
1 |
45 |
| 10 |
COCH3 |
H |
Cl |
1c
|
24 |
1 |
>99 |
| 11 |
CHO |
H |
Cl |
1c
|
24 |
1 |
60 |
| 12 |
CN |
H |
Cl |
1c
|
24 |
1 |
48 |
| 13 |
COCH3 |
H |
Br |
2a
|
3 |
1 |
>99 |
| 14 |
COCH3 |
H |
Br |
2b
|
3 |
1 |
>99 |
| 15 |
COCH3 |
H |
Br |
2c
|
3 |
1 |
>99 |
| 16 |
COCH3 |
H |
Br |
2c
|
6 |
0.5 |
>99 |
| 17 |
CHO |
H |
Br |
2c
|
8 |
1 |
>99 |
| 18 |
CN |
H |
Br |
2c
|
10 |
1 |
>99 |
| 19 |
COCH3 |
H |
Cl |
2c
|
18 |
1 |
>99 |
| 20 |
CHO |
H |
Cl |
2c
|
24 |
1 |
75 |
| 21 |
CN |
H |
Cl |
2c
|
24 |
1 |
56 |
| 22 |
COCH3 |
H |
Br |
[Ru]
|
24 |
1 |
>99 |
| 23 |
COCH3 |
H |
Br |
[Ru] +
1c
|
5.5 |
1c |
>99 |
In order to examine whether incorporation of ruthenium along with palladium in the same ligand frame has been effective in enhancing the catalytic efficiency, the heterodinuclear Pd–Ru complexes (2a, 2b and 2c) ware also tested as catalysts towards Suzuki coupling reactions, and the results are included in Table 4 (entries 13–21). Interestingly, all the three Pd–Ru complexes showed much better, and mutually comparable,12 catalytic efficiency compared to their mononuclear Pd-precursors. For example, using 1 mol% of 2c as catalyst the Suzuki coupling of p-bromoacetophenone or p-chloroacetophenone with phenylboronic acid was achieved, in relatively shorter time (entries 15 and 19), than the corresponding coupling reaction, using 1c as catalyst. Here also, reduction in catalyst loading results in an increase in reaction time for maintaining the yield (entries 15 and 16). With electron-withdrawing substituents (e.g. CHO, CN) in the aryl halides, a sharp decrease in activity (relative to COCH3 substituent), in terms of both time and yield, is observed (entries 17, 18 and 20, 21).
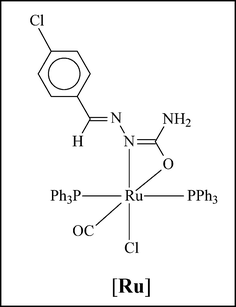
The significant enhancement of catalytic efficiency upon incorporation of ruthenium in the palladium-bound semicarbazone is a clear manifestation of cooperativity of the two different metal centers.13 The individual capacity of the palladium center has already been displayed in the catalytic activity of the mononuclear palladium complexes (1a–1c). In order to assess the individual capacity of the ruthenium center in the Pd–Ru complexes (2a–2c), we have prepared a mononuclear ruthenium complex, denoted by [Ru],3c in which the ruthenium center has a very similar coordination environment to the Pd–Ru complexes. The catalytic efficiency of this [Ru] complex has been examined towards Suzuki coupling reaction between p-bromoacetophenone and phenylboronic acid, and the complex has been found to successfully catalyze the coupling reaction (entry 22), however, with much less efficiency compared to the mononuclear Pd complexes (entry 3). Similarly, the catalytic efficiency of a 1:1 mixture of the [Ru] complex and complex 1c has been found to be significantly less (entry 23) than that of complex 2c alone (entry 15), indicating the cooperative effect of the two metal centers in the dimetallic complexes.
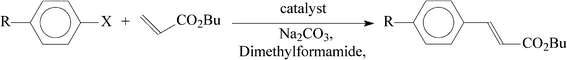
Encouraged by the facile Suzuki coupling reactions catalyzed by the Pd and Pd–Ru complexes, we have further investigated the catalytic activity of these complexes in the Heck coupling reaction between aryl halides and butyl acrylate. All three complexes of each group (mononuclear and heterodinuclear) showed comparable reactivity, and the results for complexes 1c and 2c are shown in Table 5. Compared to the Suzuki coupling reactions, the Heck coupling reactions required relatively high temperature and longer time. For example, when the reaction of p-bromoacetophenone and butyl acrylate was performed using 1c as catalyst at 120 °C, 42 h was needed to achieve only moderate yield (entry 1). However, on increasing the temperature to 145 °C, full conversion could be achieved in much less time (entry 2). Keeping the reaction temperature fixed at 145 °C, if the amount of catalyst is reduced, the time required for full conversion increases proportionally (entry 3). The time for full conversion also varies with variation of the substituent R in the aryl bromide (entries 4 and 5). When aryl chlorides were used instead of aryl bromides, a sharp decrease in yields was observed along with much longer reaction time (entries 6, 7 and 8). Using 2c as the catalyst for the same coupling reactions of aryl bromides and butyl acrylate, much less time was required to achieve similar yields (entries 9–12). In the case of the coupling of aryl chlorides and butyl acrylate, significantly higher yields were obtained with 2c as catalyst (entries 13, 14 and 15). Again, to assess the contribution of the ruthenium center, we have examined the catalytic efficiency of the [Ru] complex in the Heck coupling reaction between p-bromoacetophenone and butyl acrylate. While the [Ru] complex could successfully catalyze the coupling reaction (entry 16), the observed efficiency was much less compared to 1c (entry 2).
| Entry |
R |
X |
Catalyst |
T/°C |
Time, h |
Amt of cat., mol (%) |
Conversion,b % |
|
Reaction conditions: aryl halide (1.0 mmol), butyl acrylate (4.0 mmol), Na2CO3 (2.4 mmol), catalyst, dimethylformamide (4 mL), 145 °C.
Determined by 1H NMR on the basis of residual aryl halide.20
|
| 1 |
COCH3 |
Br |
1c
|
120 |
42 |
1 |
56 |
| 2 |
COCH3 |
Br |
1c
|
145 |
8 |
1 |
>99 |
| 3 |
COCH3 |
Br |
1c
|
145 |
16 |
0.5 |
>99 |
| 4 |
CHO |
Br |
1c
|
145 |
9 |
1 |
>99 |
| 5 |
CN |
Br |
1c
|
145 |
11 |
1 |
>99 |
| 6 |
COCH3 |
Cl |
1c
|
145 |
24 |
1 |
74 |
| 7 |
CHO |
Cl |
1c
|
145 |
24 |
1 |
52 |
| 8 |
CN |
Cl |
1c
|
145 |
24 |
1 |
37 |
| 9 |
COCH3 |
Br |
2c
|
145 |
5 |
1 |
>99 |
| 10 |
COCH3 |
Br |
2c
|
145 |
10 |
0.5 |
>99 |
| 11 |
CHO |
Br |
2c
|
145 |
7 |
1 |
>99 |
| 12 |
CN |
Br |
2c
|
145 |
10 |
1 |
>99 |
| 13 |
COCH3 |
Cl |
2c
|
145 |
24 |
1 |
>99 |
| 14 |
CHO |
Cl |
2c
|
145 |
24 |
1 |
70 |
| 15 |
CN |
Cl |
2c
|
145 |
24 |
1 |
46 |
| 16 |
COCH3 |
Br |
[Ru]
|
145 |
24 |
1 |
75 |
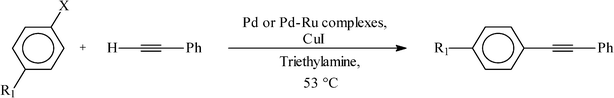
We have also scrutinized the catalytic efficiency of the six (1a, 1b, 1c and 2a, 2b, 2c) complexes in the Sonogashira coupling reaction between aryl halides and phenylacetylene. As before, all the three complexes of each family showed comparable reactivity and results, thus only 1c and 2c are presented in Table 6. In the case of aryl bromides, high yields of the coupled products were obtained at relatively low temperature (53 °C) using 1 mol% of 1c as catalyst (entries 1, 2 and 3). In the case of aryl chlorides the efficiency of the catalysts decreases significantly (entries 4, 5 and 6). Interestingly, in the Sonogashira reactions yield of the coupled products did not improve upon using 2c as catalyst instead of 1c (entries 7–12). It was also found that the [Ru] complex alone was not able to catalyze the Sonogashira coupling reaction (entry 13), which explains why incorporation of ruthenium along with palladium has not been effective in enhancing the catalytic efficiency.
| Entry |
R |
X |
Catalyst |
Time, h |
Amt of cat., mol (%) |
Conversion,b % |
|
Reaction conditions: aryl halide (1.0 mmol), phenylacetelene (1.2 mmol), CuI (10 mol%), catalyst, triethylamine (4 mL), 53 °C.
Determined by 1H NMR on the basis of residual aryl halide.20
|
| 1 |
COCH3 |
Br |
1c
|
24 |
1 |
>99 |
| 2 |
CHO |
Br |
1c
|
24 |
1 |
>99 |
| 3 |
CN |
Br |
1c
|
24 |
1 |
>99 |
| 4 |
COCH3 |
Cl |
1c
|
24 |
1 |
48 |
| 5 |
CHO |
Cl |
1c
|
24 |
1 |
37 |
| 6 |
CN |
Cl |
1c
|
24 |
1 |
31 |
| 7 |
COCH3 |
Br |
2c
|
24 |
1 |
>99 |
| 8 |
CHO |
Br |
2c
|
24 |
1 |
>99 |
| 9 |
CN |
Br |
2c
|
24 |
1 |
>99 |
| 10 |
COCH3 |
Cl |
2c
|
24 |
1 |
50 |
| 11 |
CHO |
Cl |
2c
|
24 |
1 |
43 |
| 12 |
CN |
Cl |
2c
|
24 |
1 |
35 |
| 13 |
COCH3 |
Br |
[Ru]
|
24 |
1 |
0 |
The present study thus demonstrates that the heterodinuclear Pd–Ru complexes (2a, 2b and 2c) are much more efficient catalysts, in terms of reaction condition and yield, than the corresponding mononuclear Pd complexes (1a, 1b and 1c) for the Suzuki and Heck coupling. However, both the Pd and Pd–Ru complexes have almost similar activity towards Sonogashira coupling. One notable aspect of the observed catalysis is that these complexes can efficiently catalyze the Suzuki and Heck coupling involving the aryl chlorides, which is relatively less common compared to aryl bromides or iodides.14 Another notable aspect is that no additional ligand was necessary for the cross-coupling reactions, and such ligand-free catalysis of C–C coupling reactions is also relatively less common.15
Conclusions
The present study shows that the salicylaldehyde semicarbazone and the two similar ligands (L1, L2 and L3) can smoothly bind to palladium, displaying an unusual tridentate ONN-mode of binding (VII). The resulting palladium complexes (1a, 1b and 1c) are found to be useful as building blocks for the synthesis of heterodinuclear complexes, as exemplified through isolation of three such complexes (2a, 2b and 2c) incorporating ruthenium as the second metal center. The mononuclear Pd complexes, as well as the heterodinuclear Pd–Ru complexes, are found to be excellent catalysts for promoting the C–C coupling (Suzuki, Heck and Sonogashira) reactions involving aryl chlorides and aryl bromides. The Pd–Ru complexes (2a, 2b and 2c) show higher catalytic activity than the Pd complexes (1a, 1b and 1c) towards Suzuki and Heck coupling due to cooperativity of the two metal centers. Attempts to utilize the mononuclear palladium complexes as efficient building blocks for the synthesis of heterodinuclear complexes, incorporating other metal centers (other than ruthenium), are in progress.
Experimental
General procedures
Commercial palladium chloride and ruthenium trichloride were purchased from Arora Matthey, Kolkata, India. [Pd(PPh3)2Cl2] and [Ru(PPh3)2(CO)2Cl2] were prepared by following reported procedures.16,17 Salicylaldehyde, 2-hydroxyacetophenone and 2-hydroxynaphthaldehyde were obtained from S.D. fine-chem, Mumbai, India. Semicarbazide hydrochloride was procured from Loba Chemie, Mumbai, India. Tetrabutylammoniumhexafluorophosphate (TBHP), used in electrochemical work, was purchased from Sigma-Aldrich. All other chemicals and solvents were reagent grade commercial materials and were used as received. The semicarbazone ligands (L1, L2, and L3) were prepared by condensing the respective aldehyde or ketone with semicarbazide in hot ethanol. Microanalyses (C, H, and N) were performed using a Heraeus Carlo Erba 1108 elemental analyzer. 1H NMR spectra were recorded in CDCl3 solution on a Bruker Avance DPX 300 NMR spectrometer using TMS as the internal standard. IR spectra were obtained on a Shimadzu FTIR-8300 spectrometer with samples prepared as KBr pellets. Electronic spectra were recorded on a JASCO V-570 spectrophotometer. Optimization of ground-state structures and energy calculations for all the complexes were carried out by density functional theory (DFT) method using the Gaussian 03 (B3LYP/SDD-6-31G) package.8 Electrochemical measurements were made using a CH Instruments model 600A electrochemical analyzer. A platinum disc working electrode, a platinum wire auxiliary electrode and an aqueous saturated calomel reference electrode (SCE) were used in the cyclic voltammetry experiments. All electrochemical experiments were performed under a dinitrogen atmosphere. All electrochemical data were collected at 298 K and are uncorrected for junction potentials.
Synthesis of complexes
1a.
To a solution of L1 (26 mg, 0.14 mmol) in hot ethanol (30 mL) was added triethylamine (28 mg, 0.28 mmol) followed by [Pd(PPh3)2Cl2] (100 mg, 0.14 mmol). The mixture was heated at reflux for 5 h to yield an orange solution. The solvent was evaporated, and the solid mass, thus obtained, was subjected to purification by thin-layer chromatography on a silica plate. With 1:3 acetonitrile-benzene as the eluant, a yellow band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract complex 1a was obtained as a crystalline yellow solid. Yield: 53%. Anal. calcd for C26H22O2N3PPd: C 52.49; H 3.70; N 7.06. Found: C 52.98; H 3.44; N 7.02%. 1H NMR(300 MHz, CDCl3):18δ (ppm) = 6.53 (t, J = 7.2 Hz, C5–H), 6.70 (d, J = 8.4 Hz, C6–H), 7.08 (t, J = 7.5 Hz, C4–H), 7.14 (d, J = 9.0 Hz, C3–H), 7.32–7.62 (PPh3 + azomethine H)*, 7.72 (s, N2–H), 11.10 (s, N1–H). IR (wave number, cm−1): 535, 694, 743, 1089, 1375, 1632, 1681, 3052, 3401.
1b.
This complex was synthesized by following the same above procedure using L2 instead of L1. Yield: 55%. Anal. calcd for C27H24O2N3PPd: C 54.51; H 4.20; N 7.06. Found: C 54.74; H 4.50; N 7.02%. 1H NMR(300 MHz, CDCl3): δ (ppm) = 2.32 (s, CH3), 6.64 (t, J = 7.5 Hz, C5–H), 6.76 (d, J = 9.0 Hz, C6–H), 6.97 (d, J = 9.0 Hz, C3–H), 7.12 (t, J = 7.2 Hz, C4–H), 7.43–7.71 (PPh3 + N2–H)*, 11.15 (s, N1–H). IR (wave number, cm−1): 535, 694, 742, 1085, 1372, 1628, 1678, 3055, 3406.
1c.
This complex was synthesized by following the same above procedure using L3 instead of L2. Yield: 62%. Anal. calcd for C30H24O2N3PPd: C 60.56; H 4.04; N 7.06. Found: C 60.48; H 4.02; N 7.02%. 1H NMR(300 MHz, CDCl3): δ (ppm) = 6.91 (d, J = 9.0 Hz, C5–H), 7.20 (t, J = 7.4 Hz, C6–H), 7.41 (t, J = 7.5 Hz, C7–H), 7.46–7.75 (PPh3 + C8–H + azomethine H)*, 7.63 (d, J = 7.5 Hz, C4–H), 8.02 (d, J = 8.5 Hz, C3–H), 8.83 (d, J = 8.4 Hz, N2–H), 11.97 (s, N1–H). IR (wave number, cm−1): 536, 692, 744, 821, 1097, 1184, 1334, 1396, 1429, 1639, 1683, 3053, 3404.
2a.
To a solution of 1a (50 mg, 0.09 mmol) in hot ethanol (30 mL) was added triethylamine (9 mg, 0.09 mmol) followed by [Ru(PPh3)2(CO)2Cl2] (70 mg, 0.09 mmol). The mixture was heated at reflux for 48 h whereby an orange solution was obtained, which was allowed to evaporate in air to yield an orange solid. Purification was achieved by thin-layer chromatography on a silica plate. With benzene as the eluant, an orange band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract the 2a complex was obtained as an orange crystalline solid. Yield: 43%. Anal. calcd for C63H51N3O3P3ClPdRu: C 61.35; H 4.14; N 3.41. Found: C 61.55; H 4.28; N 3.67%. 1H NMR(300 MHz, CDCl3): δ (ppm) = 7.39–7.87 (3PPh3 + C3–H to C6–H)*, 8.25 (d, J = 7.2 Hz, azomethine H), 11.22 (s, N1–H). IR (wave number, cm−1): 519, 692, 745, 1091, 1379, 1621, 1917, 3050.
2b.
This complex was synthesized by following the same above procedure using 1b instead of 1a. Yield: 42%. Anal. calcd for C64H53N3O3P3ClPdRu: C 61.54; H 4.33; N 3.41. Found: C 61.59; H 4.25; N 3.37%. 1H NMR(300 MHz, CDCl3): δ (ppm) = 2.22 (s, CH3), 7.32–7.89 (3PPh3 + C4–H to C6–H)*, 8.12 (d, J = 7.5 Hz, C3–H), 10.89 (s, N1–H). IR (wave number, cm−1): 517, 692, 743, 1088, 1381, 1619, 1921, 3051.
2c.
To a solution of 1c (50 mg, 0.08 mmol) in hot ethanol (30 mL) was added triethylamine (8 mg, 0.08 mmol) followed by [Ru(PPh3)2(CO)2Cl2] (63 mg, 0.08 mmol). The mixture was heated at reflux for 48 h. 2c separated as an orange precipitate, which was allowed to settle down. The solid was collected by filtration and washed with small amount of alcohol and dried in air. Yield: 60%. Anal. calcd for C67H55N3O3P3ClPdRu: C 62.57; H 4.28; N 3.27. Found: C 62.44; H 4.50; N 3.22%. 1H NMR(300 MHz, CDCl3): δ (ppm) = 6.91 (d, J = 5.4 Hz, C5–H), 7.01–7.95 (3PPh3 + C3–H + C4–H + C6–H to C8–H + azomethine H)*, 11.98 (s, N1–H). IR (wave number, cm−1): 515, 694, 744, 820, 1095, 1188, 1331, 1383, 1436, 1512, 1612, 1919, 3053.
Crystallography
Single crystals of 1b and 1c were obtained by slow diffusion of hexane into dichloromethane solutions of the complexes. Single crystals of 2a and 2c were obtained by slow evaporation of solvent from solutions of the complexes in acetonitrile. Selected crystal data and data collection parameters of 1b and 2a are given in Table 7, and those of 1c and 2c are given in Table S3.† Data on 1b and 1c were collected respectively on a Marresearch Image Plate system and Siemens P4 diffractometer. Data on 2a and 2c were collected on a Bruker SMART Apex CCD area detector. X-ray data reduction, structure solution and refinement were done using SHELXS-97 and SHELXL-97 programs.19 The structures were solved by direct methods.
| |
1b
|
2a
|
|
R1 = Σ||Fo| − |Fc||/Σ|Fo|.
wR2 = [Σ{w(Fo2 − Fc2)2}/Σ{w(Fo2)}]1/2.
GOF = [Σ(w(Fo2 − Fc2)2)/(M − N)]1/2, where M is the number of reflections and N is the number of parameters refined.
|
| Empirical formula |
C27H24N3O2PPd |
C63H51ClN3O3PdRu |
| Formula weight |
559.88 |
1316.03 |
| Space group |
Triclinic, P1 |
Triclinic, P1 |
|
a/Å |
10.1264(9) |
10.2779(2) |
|
b/Å |
14.1165(12) |
13.7689(2) |
|
c/Å |
16.8322(12) |
22.0341(3) |
|
α (°) |
80.414(7) |
99.315(1) |
|
β (°) |
84.706(7) |
103.302(1) |
|
γ (°) |
78.503(7) |
100.393(1) |
|
V/Å3 |
2320.6(3) |
2916.58(8) |
|
Z
|
4 |
2 |
|
λ/Å |
0.71073 |
0.748 |
| cryst size/mm3 |
0.30 × 0.05 × 0.05 |
0.22 × 0.16 × 0.14 |
|
T/K |
150 |
296 |
|
μ/mm−1 |
0.900 |
0.748 |
|
R
1
a
|
0.0436 |
0.0415 |
|
wR
2
b
|
0.0976 |
0.1404 |
| GOFc |
0.93 |
0.88 |
General procedure for Suzuki coupling reactions.
In a typical run, an oven-dried 10 mL round bottom flask was charged with a known mole percent of catalyst, base (2.4 mmol), phenylboronic acid (1.2 mmol) and aryl halide (1 mmol) with ethanol-toluene 1:1 mixture. The flask was placed in a preheated oil bath at 95 °C. After completion of the reaction (monitored by TLC), the flask was removed from the oil bath and water (20 mL) added, followed by extraction with ether (4 × 10 mL). The combined organic layers were washed with water (3 × 10 mL), dried over anhydrous Na2SO4, and filtered. Solvent was removed under vaccum. The residue was dissolved in CDCl3 and analyzed by 1H NMR. Percent conversions were determined against the remaining aryl halide.20
General procedure for Heck coupling reactions.
In a typical run, an oven-dried 10 ml round bottom flask was charged with a known mole percent of catalyst, base (2.4 mmol), n-butyl acrylate (4.0 mmol) and aryl halide (1 mmol) with dimethylformamide. The flask was placed in a preheated oil bath at 145 °C. The work-up and product analysis were done as before.
General procedure for Sonogashira coupling reactions.
To a slurry of aryl halide (1 mmol), cuprous iodide (10 mol%) and catalyst (a known mol%) in triethylamine (4 mL), phenylacetylene (1.2 mmol) was added and heated at required temperature. Rest of the procedure (the work-up and product analysis) was similar as before.
Acknowledgements
Financial assistance received from the Department of Science and Technology New Delhi, [Grant No. SR/S1/IC-29/2009] is gratefully acknowledged. The authors thank Dr Kajal K. Rajak for his kind help with the DFT studies. M. G. B. D. thanks EPSRC (UK) and the University of Reading for funds for the Image Plate System. Sayanti Datta and Dipravath Kumar Seth thank the Council of Scientific and Industrial Research, New Delhi, for their fellowship [Grant No. 09/096(0563)/2008-EMR-I and Grant No. 9/096(0511)/2006-EMR I respectively].
References
-
(a) S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef;
(b) S. B. Padhye and G. B. Kaffman, Coord. Chem. Rev., 1985, 63, 127–160 CrossRef CAS.
-
(a) F. R. Pavan, P. I. S. Maia, S. R. A. Leite, V. M. Deflon, A. A. Batista, D. N. Sato, S. G. Franzblau and C. Q. F. Leite, Eur. J. Med. Chem., 2010, 45, 1898–1905 CrossRef CAS;
(b) S. M. Sh. Atta, D. S. Farrag, A. M. K. Sweed and A.H. Abdel-Rahman, Eur. J. Med. Chem., 2010, 45, 4920–4927 CrossRef CAS;
(c) A. Chipeleme, J. Gut, P. J. Rosenthal and K. Chibale, Bioorg. Med. Chem., 2007, 15, 273–282 CrossRef CAS;
(d) N. C. Kasuga, K. Onodera, S. Nakano, K. Hayashi and K. Nomiya, J. Inorg. Biochem., 2006, 100, 1176–1186 CrossRef CAS;
(e) K. Nomiya, K. Sekino, M. Ishikawa, A. Honda, M. Yokoyama, N. C. Kasuga, H. Yokoyama, S. Nakano and K. Onoder, J. Inorg. Biochem., 2004, 98, 601–615 CrossRef CAS.
-
(a) I. Pal, S. Dutta, F. Basuli, S. Goverdhan, S. M. Peng, G. H. Lee and S. Bhattacharya, Inorg. Chem., 2003, 42, 4338–4345 CrossRef CAS;
(b) P. Gupta, F. Basuli, S. M. Peng, G. H. Lee and S. Bhattacharya, Inorg. Chem., 2003, 42, 2069–2074 CrossRef CAS;
(c) F. Basuli, S. M. Peng and S. Bhattacharya, Inorg. Chem., 2001, 40, 1126–1133 CrossRef CAS.
-
(a) S. B. Novakovic, G. A. Bogdanovic, I. D. Brceski and V. M. Leovac, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, C65, m263–m265 CAS;
(b) A. M. B. Bastos, J.G. Silva, P. I. S. Maia, V. M. Deflon, A. Batista, A. V. M. Ferreira, L.M. Botion, E. Niquet and H. Beraldo, Polyhedron, 2008, 27, 1787–1794 CrossRef CAS;
(c) G. F. Sousa, V. M. Deflon, M. T. P. Gambardella, R. H. P. Francisco, J. D. Ardisson and E. Niquet, Inorg. Chem., 2006, 45, 4518–4525 CrossRef;
(d) N.C. Kasuga, K. Onodera, S. Nakano, K. Hayashi and K. Nomiya, J. Inorg. Biochem., 2006, 100, 1176–1186 CrossRef CAS.
-
(a) V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS;
(b) C. Barnard, Platinum Met. Rev., 2008, 52, 38–45 CrossRef CAS;
(c) A. Fihri, P. Meunier and J. C. Hierso, Coord. Chem. Rev., 2007, 251, 2017–2055 CrossRef CAS;
(d) R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS;
(e) C. J. Elsevier, Coord. Chem. Rev., 1999, 185, 809–822 CrossRef.
-
(a) V. Mahalingam, N. Chitrapriya, F. R. Fronczek and K. Natarajan, Polyhedron, 2010, 29, 3363–3371 CrossRef CAS;
(b) M. R. P. Kurup, B. Varghese, M. Sithambaresan, S. Krishnan, S. R. Sheeja and E. Suresh, Polyhedron, 2010, 29, 3318–3323 CrossRef;
(c) M. Safavi, A. Foroumadi, M. Nakhjiri, M. Abdollahi, A. Shafiee, H. Ilkhani, M. R. Ganjali and S. Emami, Bioorg. Med. Chem. Lett., 2010, 20, 3070–3073 CrossRef CAS;
(d) V. V. Bon, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m300–m302 Search PubMed;
(e) S. Bjelogrlic, T. Todorovic, A. Bacchi, M. Zec, D. Sladic, T. Srdic-Rajic, D. Radanovic and K. Andelkovic, J. Inorg. Biochem., 2010, 104, 673–682 CrossRef CAS.
-
(a) N. Gomez-Blanco, J. J. Fernandez, A. Fernandez, D. Fernandez, M. Lopez-Torres and J.M. Vila, Eur. J. Inorg. Chem., 2009, 3071–3083 CrossRef CAS;
(b) S. Halder, S. M. Peng, G. H. Lee, T. Chatterjee, A. Mukherjee, S. Dutta, U. Sanyal and S. Bhattacharya, New J. Chem., 2008, 32, 105–114 RSC;
(c) S. Halder, M. G. B. Drew and S. Bhattacharya, J. Chem. Sci., 2008, 120, 441–446 CrossRef CAS;
(d) I. Angurell, I. Martinez-Ruiz, O. Rossell, M. Seco, P. Gomez-Sal, A. Martin, M. Font-Bardia and X. Solans, J. Organomet. Chem., 2007, 692, 3882–3891 CrossRef CAS.
-
M. J. Frisch, G. W. Tracks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman Jr, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Calamani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Ukuda, I. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskroz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian 03, revision D01; Gaussian Inc.: Pittsburgh, PA., 2003 Search PubMed.
- Phenyl rings of the triphenylphosphines have been replaced by hydrogens.
- Contribution from semicarbazone in the dinuclear complexes is much less (particularly in LUMO) compared to that in the corresponding mononuclear complexes.
- A little dichloromethane was necessary to take the complex into solution. Addition of a large
excess of acetonitrile was necessary to record the redox responses in proper shape.
- Results obtained for coupling of p-bromoacetophenone with phenylboronic acid are cited only.
-
(a) D. C. Powers and T. Ritter, Acc. Chem. Res., 2011 DOI:10.1021/ar2001974 ASAP;
(b) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS;
(c) R. D. Adams and B. Captain, Acc. Chem. Res., 2009, 42, 409–418 CrossRef CAS;
(d) C. Wang and Z. Xi, Chem. Soc. Rev., 2007, 36, 1395–1406 RSC;
(e)
M. Shibasaki, Y. Yamamoto, Multimetallic Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed;
(f)
B. Cornils, W. A. Herrmann, R. Schlögl and C.-H. Wong, Catalysis from A to Z, Wiley-VCH, Weinheim, 2003 Search PubMed;
(g)
R. A. Adams, F. A. Cotton, Catalysis by Di- and Polynuclear Metal Cluster Complexes, Wiley-VCH, Weinheim, 1998 Search PubMed;
(h) J. Xiao and R. J. Puddephatt, Coord. Chem. Rev., 1995, 143, 457–500 CrossRef CAS.
-
(a) F. Godoy, C. Segarra, M. Poyatos and E. Peris, Organometallics, 2011, 30, 684–688 CrossRef CAS;
(b) R. K. Das, B. Saha, S. M. W. Rahaman and J. K. Bera, Chem.–Eur. J., 2010, 16, 14459–14468 CrossRef CAS;
(c) H. Firouzabadi, N. Iranpoor and M. Gholinejad, J. Mol. Catal. A: Chem., 2010, 321, 110–116 CrossRef CAS;
(d) C. F. Fu, C. C. Lee, Y. H. Liu, S. M. Peng, S. Warsink, C. J. Elsevier, J. T. Chen and S. T. Liu, Inorg. Chem., 2010, 49, 3011–3018 CrossRef CAS;
(e) M. C. Lipke, R. A. Woloszynek, L. Ma and J. D. Protasiewicz, Organometallics, 2009, 28, 188–196 CrossRef CAS.
-
(a) S. M. Islam, P. Mondal, K. Tuhina, A. S. Roy, S. Mondal and D. Hossain, J. Inorg. Organomet. Polym. Mater., 2010, 20, 264–277 CrossRef CAS;
(b) M. Guo, Z. Zhu, H. Huang and Q. Zhang, Catal. Commun., 2009, 10, 865–867 CrossRef CAS;
(c) C. Pan, M. Liu, L. Zhang, H. Wu, J. Ding and J. Cheng, Catal. Commun., 2007, 9, 508–510 CrossRef.
-
H. L. Grube, in Handbook of Preparative Inorganic Chemistry, ed. G. Brauer,, Academic Press, London, 2nd edn, 1965, vol. 2, p. 1584 Search PubMed.
- N. Ahmed, S. D. Robinson and M. F. Uttley, J. Chem. Soc., Dalton Trans., 1972, 843–847 RSC.
- Overlapping signals are marked with an asterisk.
-
G. M. Sheldrick, SHELXS-97 and SHELXL-97, Fortran programs for crystal structure solution and refinement; University of Gottingen, Gottingen, Germany, 1997 Search PubMed.
-
(a) P. Evans, P. Hogg, R. Grigg, M. Nurnabi, J. Hinsley, V. Sridharan, S. Suganthan, S. Korn, S. Collard and J. E. Muir, Tetrahedron, 2005, 61, 9696–9704 CrossRef CAS;
(b) C. L. Chen, Y. H. Liu, S. H. Peng and S. T. Liu, Organometallics, 2005, 24, 1075–1081 CrossRef CAS;
(c) E. Peris, J. Mata, J. A. Loch and R. H. Crabtree, Chem. Commun., 2001, 201–202 Search PubMed;
(d) D. A. Albisson, R. B. Bedford, S.
P. Noelle and S. E. Lawrence, Chem. Commun., 1998,(19), 2095–2096 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: View of the 1c complex (Fig. S1), selected bond parameters for complexes 1c and 2c (Table S1), DFT optimized structure of 1a (Fig. S2), selected bond parameters for optimized structures of 1a and 2b (Table S2), view of the 2c complex (Fig. S3), DFT optimized structure of 2b (Fig. S4), crystallographic data for complexes 1c and 2c (Table S3). CCDC reference numbers 848667–848670. For ESI see DOI: 10.1039/c2ra20105d |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.