Modulating enzymatic activity in the presence of gold nanoparticles†
Jashmini
Deka
a,
Anumita
Paul
*a and
Arun
Chattopadhyay
*ab
aDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati, Assam, India 781 039. E-mail: arun@iitg.ernet.in and anumita@iitg.ernet.in
bCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati, Assam, India 781 039
First published on 8th March 2012
Abstract
The presence of citrate-stabilized gold nanoparticles (cit-Au NPs) significantly enhanced the specific activity of α-amylase, which could also be modulated by varying the concentration of the enzyme, while keeping the NP concentration constant. Also, the lowest concentration (0.175 μg mL−1) at which the Michaelis–Menten behavior of the enzyme could clearly be observed in the presence of NPs was much less than that in their absence. At higher protein concentrations the activity decreased monotonically until it reached nearly the same as that of free enzyme, while exhibiting Michaelis–Menten kinetics at all concentrations. Transmission electron microscopy (TEM) and dynamic light scattering (DLS) based particle size analyses indicated increased agglomeration of the NPs with increased protein concentration. The results have been explained based on a model which considered the presence of enzyme bound to NP and that available for enhanced catalysis, enzyme bound to NP but unavailable due to being buried inside the agglomerate and the free enzyme.
Introduction
Interaction of metal nanoparticles (NPs) such as those of Au (and magnetic NPs) and biomolecules like proteins, peptides, RNA and DNA provides diagnostic tools for the identification,1–3 assay and probe for the function of the molecule.4,5 Judiciously functionalized Au NPs have been extensively used not only for the detection of various enzymes but also for measurement of their activity.6–12 The high extinction coefficient and additional optical properties based on the distance of separation between two or more Au NPs make them desirable for such applications. Further, the presence of such NPs might also lead to altered activity of the biomolecule. For example, chemical or electrostatic attachment of enzymes to functionalized Au NPs may alter the catalytic properties either by increasing or decreasing the affinity for enzyme-substrate formation;13–17 enhancing its stability;13,14,18–20 enhancing the rate of product formation (although significant report in this regard is still lacking)13,14 or retention21–23 and even some loss of activity of the enzyme.18,19,24 Similarly, interaction with Au NPs has been found to enhance the stability of peptides25 and DNA.26 Thus, although there are reports on NP induced changes in the properties of enzymes, the paucity of literature is apparent especially with respect to clear exhibition of a significant difference in properties following attachment of NPs to the enzyme and understanding of the mechanisms involved. Understanding the mechanism of the NP induced alteration of the properties of biomolecules is not only central to taking full advantage of nanoscale materials but also important in knowing the implications of manufactured nanomaterials on human health and the environment.27 Further, enzymes, constituting a bulk of biomolecules, are responsible for the key functions of living systems and their utility in the catalytic production of medicinally important compounds provide a clear opportunity to study their behavior in the presence of inorganic NPs such as those of Au. Also, it would be of significance if the enzymatic kinetics were to depend not only on the presence of Au NPs but also on their concentration ratio in the medium in such a way so as to be able to modulate the kinetics. In other words, a pertinent question can be asked: would the ratio of enzyme to Au NPs change the structure of the enzyme to influence its kinetics? Addressing a part of the question would contribute towards better understanding of the fate of Au NPs if present in the human body for treatment or otherwise.Herein, we report the modulation of enzymatic activity in the presence of cit-Au NPs, where as high as a 9-fold increase in the specific activity of the digestive enzyme α-amylase in the presence of cit-Au NPs could be observed. Starch was used as the substrate for probing the enzymatic activity in the presence of cit-Au NPs. Fluorescence spectroscopic measurements and other chemical tests were performed to establish that the enzymes were attached to the NPs. Also, the activity could be modulated by merely varying the enzyme![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cit-Au NP ratio. We also propose a model for explaining our observations based on Michaelis–Menten kinetics and in terms of attachment of enzyme to the NPs and subsequent agglomeration. Transmission electron microscopic (TEM) and dynamic light scattering (DLS) based analyses were carried out to correlate the state of the NPs to the modulating activity of the enzyme in varying enzyme:cit-Au NP ratios. Different levels of agglomeration of NPs was found to be the reason behind the tuning of the enzyme's specific activity. The free thiol groups of the enzyme were blocked by 2,2′-dithiobis (5-nitropyridine) (DTNP) to carry out control experiments in order to establish their role in the favourable orientation—while attached to the NPs––and hence enhancement of the activity of the enzyme.
:cit-Au NP ratio. We also propose a model for explaining our observations based on Michaelis–Menten kinetics and in terms of attachment of enzyme to the NPs and subsequent agglomeration. Transmission electron microscopic (TEM) and dynamic light scattering (DLS) based analyses were carried out to correlate the state of the NPs to the modulating activity of the enzyme in varying enzyme:cit-Au NP ratios. Different levels of agglomeration of NPs was found to be the reason behind the tuning of the enzyme's specific activity. The free thiol groups of the enzyme were blocked by 2,2′-dithiobis (5-nitropyridine) (DTNP) to carry out control experiments in order to establish their role in the favourable orientation—while attached to the NPs––and hence enhancement of the activity of the enzyme.
Results and discussion
Cit-Au NPs were synthesized using a conventional method that resulted in an average particle size of 11 ± 2 nm. The enzymatic reactions were carried out at 37 °C using starch as the substrate in pH 7.0 buffer; by first mixing the enzyme with NPs followed by subsequent transfer to the reaction medium. For kinetic studies, the product formation was followed by maltose estimation using the well-known DNS acid method,28 details of which are available in the Experimental Section. It is worth mentioning here that control experiments were performed to rule out any possibility of interference from the cit-Au NPs in the maltose estimation test for following enzyme kinetics. The details are discussed in the Experimental Section. The experiments suggested that DNS test could be performed even in the presence of cit-Au NPs, the details of which are available in the Electronic Supplementary Information, ESI (Fig. S1†). Fig. 1 shows specific activity versus substrate concentration plots in the presence of different concentrations of enzyme while that of cit-Au NPs (i.e. 2 × diluted cit-Au NPs containing 4.0 × 1012 NPs in 5.0 mL reaction mixture, refer to ESI† for details of calculation) was kept constant (except for the control experiment performed with enzyme only). The data in each graph represent average values of three measurements. The individual graphs with error bars are shown in Fig. S2.† It is interesting to note that the results could be fitted to the Michaelis–Menten equation at and above 0.175 μg mL−1 of enzyme concentration and the calculated apparent Michaelis constant (Kam), rate constants for product formation (kacat) and maximum velocity (Vamax) values are included in Table 1. On the other hand, although at 0.087 μg mL−1 of enzyme concentration (in the presence of Au NPs) the enzyme activity was observed (refer to Fig. S2†) the data could not be fitted to the Michaelis–Menten equation and thus values as above were not calculated. As is clear from the graphs and the table, at the lowest concentration of the enzyme (0.175 μg mL−1)—where the Michaelis–Menten behavior could clearly be observed—the activity was the highest as represented by characteristic values, which were substantially different from those in the absence of the NPs. For example, the Kam value was about 0.5 times and Vamax and kacat were about 9 and 5 times respectively of that of the pure enzyme, indicating significant enhancement of both in substrate affinity and rate of product formation. Further, Kam values for higher concentrations of enzyme increased initially, leading to a constant value, while those of kacat and Vamax decreased monotonically. The lower values of the Michaelis constant (Km) at lower enzyme concentrations indicated higher affinity towards the substrate, possibly due to more favorable orientation of the enzyme structure on attaching to the cit-Au NPs. It is important to mention here that while at the lowest concentration (0.175 μg mL−1) of enzyme the specific activities were the highest, however, at the same concentration of pure enzyme the specific activities at different substrate concentrations were low and did not clearly exhibit the Michaelis–Menten behavior (refer to Fig. 2). Further experiments revealed that lowering the concentration of Au NPs (i.e. 3× and 4× diluted cit-Au NPs containing 2.7 × 1012 and 2.0 × 1012 NPs per 5.0 mL of reaction mixture respectively) did not change the values of the highest activity of the enzyme, substrate affinity or rate of product formation (refer to ESI, Fig. S3†). However at a higher concentration of cit-Au NPs (i.e. as prepared cit-Au NPs having 8.0 × 1012 NPs per 5.0 mL of reaction mixture) the Vmax and kcat were found to be half of that for 2× diluted cit-Au NPs. This could be due to the pH of the undiluted cit-Au NPs solution which was 6.4, and which is less than the optimum pH for the activity of α-amylase. In addition, at higher Au NP to enzyme ratios the agglomeration of the complex could be higher, reducing the activity of the enzyme.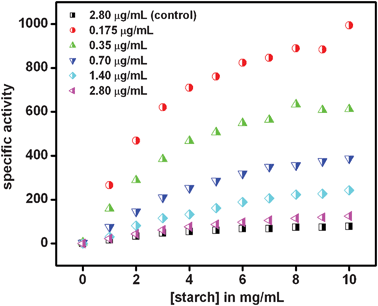 | ||
| Fig. 1 Specific activity of α-amylase (μmol mg−1 min−1) in the presence of cit-Au NPs as compared to the control (i.e. enzyme in absence of cit-Au NPs). Legend represents the respective concentration of enzyme at which the experiments were carried out. The concentration of cit-Au NPs was same for all of the above. | ||
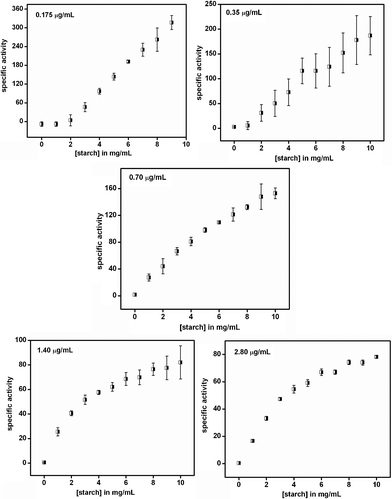 | ||
| Fig. 2 Specific activity of α-amylase (μmol mg−1 min−1) in the absence of cit-Au NPs. Legend represents the respective concentration of enzyme at which the experiments were carried out. The results are the average of three sets. | ||
| [α-amylase] (μg mL−1) | Vamax (μmol mg−1 min−1) | Kam (mg mL−1) | kacat (103 min−1) |
|---|---|---|---|
| a The data in the last row represent values corresponding to enzyme in the absence of cit-Au NPs. | |||
| 0.175 | 14.22 ± 1.54 | 4.11 ± 0.34 | 7.11 ± 0.77 |
| 0.35 | 10.00 ± 0.00 | 5.00 ± 0.00 | 5.00 ± 0.00 |
| 0.70 | 9.23 ± 1.33 | 10.00 ± 0.00 | 4.62 ± 0.66 |
| 1.40 | 8.00 ± 0.00 | 10.00 ± 0.00 | 4.00 ± 0.00 |
| 2.80 | 2.67 ± 0.00 | 10.00 ± 0.00 | 1.33 ± 0.00 |
| 2.80a | 1.67 ± 0.33 | 10.00 ± 0.00 | 1.44 ± 0.38 |
TEM measurements (Fig. 3) indicated that at the lowest concentration of the enzyme used (0.175 μg mL−1), cit-Au NPs remained isolated, while agglomeration could be observed at higher concentrations of the protein. For example, at 0.35 μg mL−1 of the protein the NPs were linearly assembled with occasional branching. On the other hand, at 2.80 μg mL−1 large agglomerated clusters could be observed. The results indicated that proteins were attached to the NPs and at higher concentrations agglomeration of the composite took place. Further, as the TEM samples were prepared following digestion of starch, the results indicated that the structures of enz–Au NP composites were stable during the process of digestion of starch. Particle size distribution analysis by DLS method (refer to ESI, Fig. S4† for graphs) also indicated increasing agglomeration of the cit-Au NPs in the presence of an increasing amount of the enzyme. For example, the hydrodynamic diameter of the cit-Au NPs in buffer was found to be 55 nm. However, in the presence of 0.175 μg mL−1 of the protein, the average hydrodynamic diameter changed to 72 ± 2 nm. It is plausible that citrate ions in the cit-Au NPs could have been replaced at least partially by the enzyme. This subsequently led to agglomeration of the NPs and the proteins (although minimal) making the average particle sizes larger. Further, as has been observed in the TEM images (Fig. 3), fusion of the NPs did not take place. On the other hand, protein–Au NP agglomerates were formed with the intervening proteins preventing the NPs from fusion. Further increase in the protein amount (0.35 μg mL−1) led to an increase in the average diameter of the NPs (84 ± 3 nm), supporting enhanced agglomeration as has also been observed by TEM. Finally, the average size of the NPs was found to be 101 ± 4 nm on increasing the protein concentration to 2.80 μg mL−1, which is again a consequence of a higher extent of agglomeration of NPs as was also evident from the TEM images. Thus the DLS results were found to support the TEM observations. Closer analyses of the DLS results (Fig. S4†) indicated that not only did the mean particle size increase but also the larger agglomerates were formed at the expense of smaller particles. In other words, the systematic shift in the average particle size indicated preferential agglomeration of the smaller particles into larger associated structures. In addition, even larger agglomerates were formed which had sizes more than an order of magnitude higher than the original sizes. The zeta potential of cit-Au NPs only in buffer showed a value of −35.6 mV, indicating the presence of a stable colloid in the medium. On the other hand, the values for cit-Au NPs in the presence of 0.175 μg mL−1, 0.35 μg mL−1 and 2.80 μg mL−1 proteins were −30.9 mV, −30.7 mV and −23.2 mV respectively. Particles with zeta potential values more positive than +30.0 mV or more negative than −30.0 mV are normally considered stable. Thus the zeta potential values suggested decreasing stability of the cit-Au NP colloids with increasing concentration of protein in the medium. Further, the magnitudes of the zeta potentials indicated partial destabilization of the NPs in the presence of protein rather than a case of complete destabilization of the colloids. In other words, it is possible that a fraction of the cit-Au NPs formed agglomerated structures in the presence of the proteins, whereas a significant portion of the same remained stable in the medium without agglomeration. This was also evident from the DLS results measured even at higher protein concentrations. Fluorescence spectroscopy studies revealed that at all concentrations, the proteins were attached to the NPs (Fig. 4), as is evident from the decrease in the intensities. As observed earlier, the presence of cit-Au NPs leads to quenching of tryptophan fluorescence, hence leading to vanishing of the fluorescence peak of α-amylase when attached to the NPs.29 In the present case the peak didn't vanish completely at higher concentrations (except for the lowest concentration of the enzyme used i.e. at 0.175 μg mL−1), indicating the presence of free enzyme in the medium in addition to those attached to the NPs. The complete disappearance of fluorescence at the lowest concentration of the enzyme means a possible absence of free enzyme in the medium at that concentration.
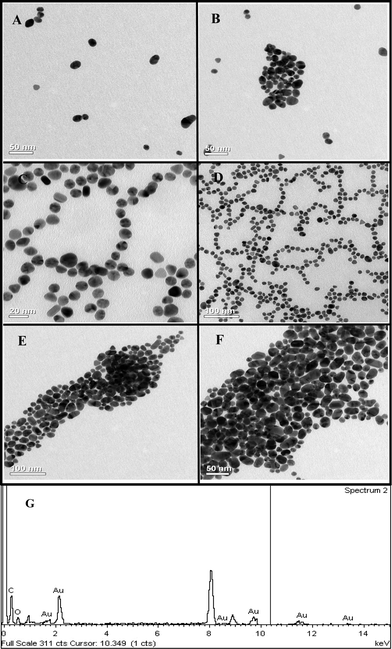 | ||
| Fig. 3 TEM images showing arrangement of Au NPs in the presence of (A and B) 0.175 μg mL−1, (C and D) 0.35 μg mL−1 and (E and F) 2.80 μg mL−1 of α-amylase in the reaction mixture post starch digestion and (G) EDX of one of the samples. | ||
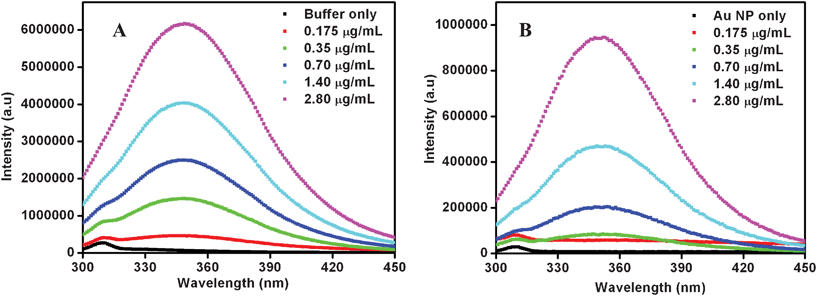 | ||
| Fig. 4 Fluorescence spectra of α-amylase in (A) absence and (B) presence of cit-Au NPs. The legends show the various concentrations of the enzyme. | ||
To our knowledge this is the first demonstration of the modulation of enzymatic activity in the presence of Au NPs based on the level of agglomeration of the NPs, which possibly controls the orientation and availability of the active site of the enzyme. Previously reported works on the modulation of an enzyme's activity in the presence of Au NPs were either dependent on the degree of enzyme–substrate association controlled by the NPs16,17 or on the size of the NPs.30 For example, You et al. reported the modulation of the catalytic behaviour of α-chymotrypsin (ChT) towards cationic, neutral, and anionic substrates in the presence of amino acid-functionalized Au clusters, which was based on the monolayer controlled uptake of the substrate and diffusion of the products through electrostatic interactions.16 In yet another report, Jordan et al. demonstrated modulation of substrate selectivity of ChT in the presence of tetraethylene glycol functionalized Au NPs (AuTEG), where interactions of AuTEG with the substrate lead to macromolecular crowding and played a major role in tuning the enzyme–substrate affinity.17
Our findings herein could be explained in terms of attachment of the enzyme to the NPs and subsequent agglomeration at higher concentrations as represented schematically in Scheme 1. α-Amylase consists of two cysteine thiol groups which are exposed to the medium and away from the active site.23 Thus, these groups could help the protein attach to the cit-Au NPs efficiently with the active site still being exposed to the medium and available for catalysis. At the lowest concentration, all the proteins were attached to the NPs and no agglomeration occurred. Because of their orientation and organization in the NPs they not only retain activity but the same was enhanced. For example, it has been recently reported that activity of lipase depends on its orientation on the functionalized Au NP surface, although no enhancement in activity was observed.31 Herein, at higher concentrations of the enzyme, agglomeration of the Au NP–enzyme mixture occurred, which increased with increasing concentration. In the agglomerated structures the enzymes which remain buried inside were no more ‘available’ for catalysis, hence the overall reaction rate decreased, although the sites which were exposed to the medium still contributed to the activity. On the other hand, there may be free enzymes present in the medium which also contributed to activity. Thus, the overall activity is the sum of that due to enzymes attached to NPs and exposed to the medium plus that due to free enzymes. In the presence of cit-Au NPs the catalytic activity of α-amylase could be represented by the product formation rate as in eqn (1)i.e., in terms of product formation from two separate but parallel reactions—one due to active enz–Au NP composites and the other due to free enzyme. Thus overall specific activity could be written in terms of specific activity of the active composite (with the mole fraction of the enzyme being x) and free enzyme (the mole fraction being y) and as written in eqn (2), with each following Michaelis–Menten behavior.
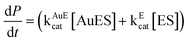 | (1) |
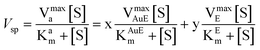 | (2) |
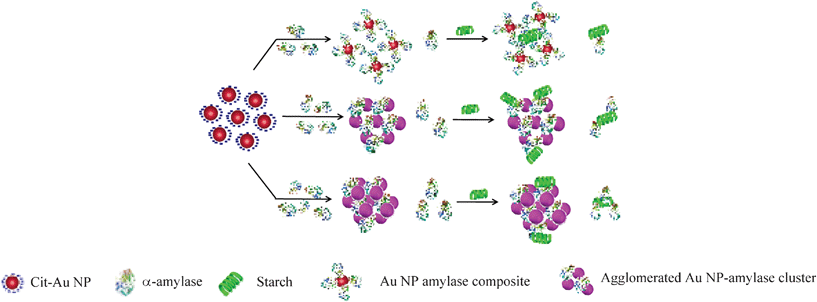 | ||
| Scheme 1 A schematic representation of possible structures present in the media consisting of cit-Au NPs and α-amylase at different enzyme concentrations. | ||
Here AuE represents the enz–Au NP active composite and E represents free enzyme. In addition, kAuEcat and kEcat are the rate constants for product formation from [AuES] and [ES] complexes respectively. Also, Vsp is the specific activity of the enzyme, Vmaxa is the apparent velocity maximum, Kam is the apparent Michaelis–Menten constant, VmaxAuE and VmaxE are the velocity maxima of the reaction for AuE and free enzyme respectively and KAuEm and KEm are the Michaelis–Menten constants for AuE and free enzyme respectively. Also, since x and y are the mole fractions of enzyme in the active composite and free enzyme present in the medium respectively z(1 − x − y) is the fraction of enzyme attached to the cit-Au NPs which remains inactive. Now, the inactive enzymes, although attached to the NPs but buried inside agglomerated structures, do not contribute to product formation. In order to account for the apparent specific activity (Fig. 1) in terms of specific activities of the active composite and the free enzyme (eqn (2)), one needs to know the values of x, y and z. This was pursued by first finding out the fraction of free enzyme. The enzyme–Au NP mixtures, with different concentrations of the enzyme, were centrifuged to precipitate and thus separate the enzyme attached to the NPs. The enzyme present in the supernatant was estimated using a method established in our laboratory.21 The different fractions of the free enzyme present in the media thus estimated are given in Table 2. Further, it was observed that at the lowest enzyme concentration used here, there was no free enzyme and no apparent agglomeration (as was observed in TEM in Fig. 3). Hence, specific activity at the lowest concentration was taken as the specific activity of the active enz–Au NP composite. Also, the specific activity of only enzyme was measured. Using these two values of specific activities and the knowledge of mole fractions of free enzyme, the observed apparent substrate-dependent specific activities at various concentrations of the enzyme, as shown in the graphs in Fig. 1, were fitted based on eqn (2). The fitted graphs are shown in Fig. 5.
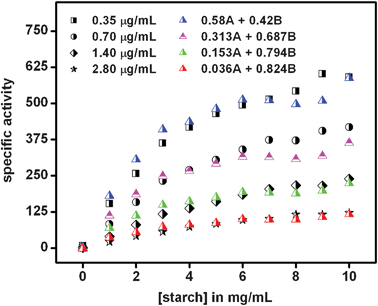 | ||
| Fig. 5 Fitting of specific activity (μmol mg−1 min−1) graphs in the presence of varying enzyme concentrations in Fig. 1 based on eqn (2). For each graph, the value of the mole fraction of active enzyme (x) attached to NPs was taken to be that which generated the best fit. The mole fraction of free enzyme (y) was found from experiments described above. Here, A represents the specific activity of the enzyme–Au NP mixture at the lowest enzyme concentration, while B is the specific activity of free enzyme. | ||
It must be mentioned here that the mole fractions of the active enzyme attached to cit-Au NP (x) were chosen such that the values resulted in the best fits of the experimental graphs (Fig. 1) using eqn (2), along with prior knowledge of specific activities and the mole fraction of the free enzyme. The values corresponding to the best fits are shown in Table 2. Obviously, the mole fractions (z) of enzyme attached to NPs but inactive could easily be calculated, once x and y are known. The results indicated that with increasing concentrations of the enzyme the fractions of inactive (z) and free (y) enzymes increased, while that of active (x) enzyme decreased, hence the observed decrease in activity. At the highest concentrations very little of the activated enzyme remained, possibly due to large-scale agglomeration and thus the overall activity was close to that of the free enzyme. As mentioned before, α-amylase contains two cysteine thiol groups which are exposed to the medium and away from the active site of the protein. This provides an additional affinity for the protein to attach to the NP via a Au–S bond, while maintaining the availability of the active site. In order to establish the role of the two free thiol groups present in α-amylase in the attachment of the protein to cit-Au NPs and thus leading to modified enzymatic activity, the thiol groups were first blocked by the treatment of the enzyme with 2,2′-dithiobis (5-nitropyridine) (DTNP). DTNP is known to modify free thiol groups in a molecule and has been used to block thiols in proteins.23,32 The details of the experimental procedure are discussed in the Experimental Section. The UV-vis spectrum of the initial mixture of α-amylase and DTNP (at 0 h of stirring) consisted of peaks at 270 nm and 319 nm corresponding to the enzyme and DTNP respectively, while that after 30 h of stirring consisted of a single peak at 387 nm, characteristic of the formation of 5-nitropyridine-2-thione (being present in the medium) as shown in Fig. 6A. This indicates the modification of the free thiol groups of the enzyme as established before.23,32 The protein was then purified by dialysis upon which the peak at 387 nm disappeared, while the peak at 270 nm due to the protein was discernible (Fig. 6A). This was followed by mixing of 950.0 μL of the modified enzyme solution to 24.0 mL of 2× cit-Au NP solution, incubating for 10 min and then used for the kinetic study as for regular experiments. The concentration of enzyme was 0.175 μg mL−1 in the final reaction mixture. For comparison, two more control experiments were run in parallel with 0.175 μg mL−1 of unmodified enzyme in the presence and absence of cit-Au NPs. The Vmax, kcat and Km were calculated as discussed earlier. The Vmax, kcat and Km for the unmodified enzyme in the presence of cit-Au NPs were found to be 16.00 × 102 μmol mg−1 min−1, 8.00 × 104 min−1 and 5.0 mg mL−1 respectively which are similar to the values obtained earlier with this concentration of enzyme. However, with the same amount of modified enzyme, the specific activities at different substrate concentrations were lower than the unmodified enzyme as shown in Fig. 6B. Moreover, the data for the modified enzyme did not clearly exhibit Michaelis–Menten behavior and thus the Vmax, kcat and Km values for the above were not calculated. It is interesting to note that the control experiment with free enzyme (at 0.175 μg mL−1) in the absence of cit-Au NPs provided similar results as that for the modified enzyme in the presence of cit-Au NPs (Fig. 6B). From these observations it can be concluded that after modification of the free and exposed thiol groups in α-amylase, the enzyme didn't exhibit any enhancement in its specific activity in the presence of cit-Au NPs and may have behaved like a free enzyme or might have been attached to the NPs randomly leading to loss of activity. These results indicated that the free thiol groups played a crucial role in the attachment of α-amylase to the cit-Au NPs, hence attaining a favorable orientation for better activity. As mentioned earlier, it is plausible that at lower concentrations the enzymes bind to the Au NPs such that the free and exposed thiol groups are attached to the NPs, while the active sites remain exposed to the medium and are available for catalytic activity. This might provide two advantages. Firstly, a collection of neighbouring protein molecules attached to the same NP could digest the same starch molecule with enhanced reactivity—owing to their favorable orientation and being able to digest the same molecule simultaneously and thus increasing kcat. Secondly, the favourable attachment and being together could lead to better enzyme–substrate complex formation, thus lowering the Michaelis–Menten constant.
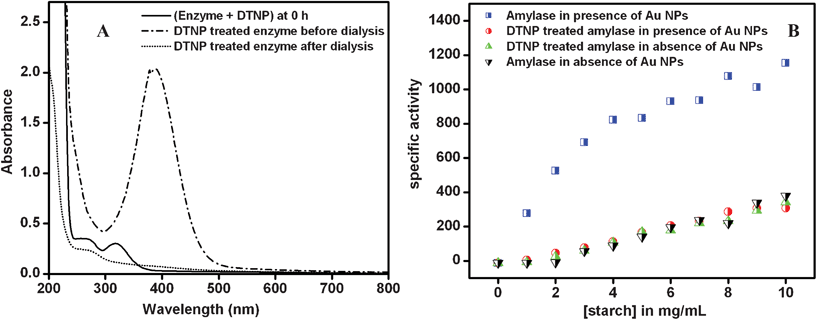 | ||
| Fig. 6 (A) UV-vis spectra of the reaction mixture of DTNP and enzyme at 0 h and after 30 h of reaction and of the same after dialysis; (B) specific activity (in μmol mg−1 min−1) of DTNP modified α-amylase in the presence of cit-Au NPs as compared to the controls (i.e. unmodified enzyme in the absence and presence of cit-Au NPs). | ||
On the other hand, when the thiol groups were blocked it may be that the proteins were attached to the NP randomly without any favorable orientation for enhanced enzyme activity. Also, the UV-vis spectrum of cit-Au NPs in the presence of modified α-amylase (refer to Fig. S5, ESI†) show noticeable peak broadening suggesting formation of agglomerated protein-clusters. These protein–NP agglomerated structures would also lead to loss of activity of the attached proteins. Thus, essentially the remainder of the free proteins would provide activity, the concentration of which might not have been sufficiently high for measurable activity. It must be mentioned here that the concentration of modified protein used was low (0.175 μg mL−1). At this concentration, unmodified protein didn't exhibit clear Michaelis–Menten kinetic behavior. In this case of modified protein it can be anticipated that some of those proteins in the medium would be attached to Au NPs in the agglomerates. Thus, this will make the concentration of free protein even lower. The observed reaction rate was nearly the same as that of free protein. Thus proteins attached to Au NPs didn't contribute to any increase in the overall rate of the reaction, which may be due to unfavorable attachment to NPs and subsequent agglomeration. A schematic representation of the two possible scenarios of enzyme activities of the unmodified and thiol group blocked enzymes in the presence of Au NPs and based on the above discussion, is shown in Scheme 2.
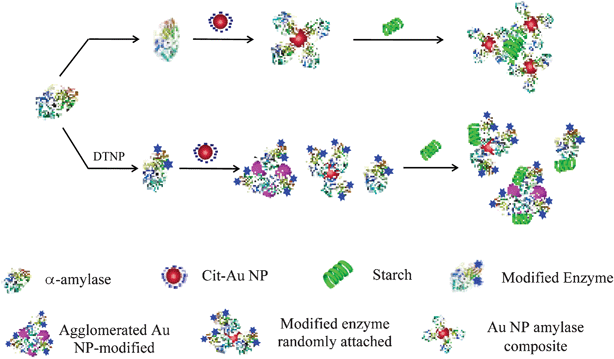 | ||
| Scheme 2 A schematic representation showing the effect of blocking the free thiol groups of the enzyme in its starch digestion in the presence of cit-Au NPs. The blue colored stars in the DTNP modified enzyme indicates blocked thiol groups. | ||
Further, in order to check the stability of the enz–Au NP agglomerated clusters, additional cit-Au NPs (equivalent to one fourth of the cit-Au NPs used for experiments) were added to the medium containing 2.80 μg mL−1 enzyme and cit-Au NPs and the substrate concentration dependent specific activities were measured. The results are shown in Fig. S6† of the ESI, which yielded Vmax, kcat and Km values equal to 4.00 × 102 μmol mg−1 min−1, 2.00 × 104 min−1 and 10.00 mg mL−1 respectively, representing slightly higher activity than in absence of excess NPs.
Also, TEM images of the enzyme–Au NP mixture in the presence of additional cit-Au NPs are shown in Fig. S7† of the ESI. The presence of smaller Au NP clusters was observed, indicating possible formation of additional agglomerates with free enzyme that was present in the medium. This possibly contributed to a small increase in activity.
Conclusion
In brief, through a series of experiments we have been able to demonstrate that details of the structures of the composite of enzymes and cit-Au NPs, when attached to each other, are important in determining the overall activity of the enzyme. For the enzyme used here (α-amylase) the exposed thiol groups present in the structure were found to be responsible for the attachment to cit-Au NPs and to possibly have better orientation for enhanced reactivity. However, occurrence and the extent of aggregation at higher enzyme concentrations determined the overall activity with increasing agglomeration leading to decreased activity. It is plausible that a group of proteins attached simultaneously to a single NP, with their active sites exposed to the medium, which may have led to cooperative digestion of the same starch molecule and be one of the reasons behind the enhanced activity. In the agglomerates, active sites of the enzymes are not accessible to starch molecules (even in the case of retention of native structure) thereby effectively lowering the activity of the enzyme. One cannot also discount the possibility of denaturation of the enzyme in the agglomerated structure and hence a reduction of activity. It may also be mentioned here that previous studies in our laboratory21 and others have indicated that interaction of Au NPs and proteins may lead to agglomeration. The origin of agglomeration could be due to electrostatic or van der Waals interactions between the NP and the protein. On the other hand, changes in the conformation of protein upon attachment to the NP may lead to agglomeration with additional proteins being present in the vicinity. Finally, cumulative effects of all of the above interactions might play roles in the overall agglomeration of proteins and NP.33,34 That the activity of enzymes in the presence of cit-Au NPs depended on the ratio of enzyme to Au NPs provided a new way of modulating enzymatic activity in the presence of NPs. The results presented here may provoke further studies involving interactions between NPs and biomolecules, essential in understanding the consequences of the use of manufactured nanomaterials in human health and environmental systems, in addition to being useful in catalysis.Experimental section
Preparation of citrate stabilized Au NPs
750.0 μL of 1.73 × 10−2 M HAuCl4 (obtained from Sigma-Aldrich Chemical Co.) was added to 30.0 mL of MilliQ grade water and then heated to boiling. Then 126.0 mg of trisodium citrate 2-hydrate (procured from E. Merck (India) Limited, Mumbai) being dissolved in 1.0 mL MilliQ grade water was added to the boiling solution (all at once) and refluxed for another 30 min to ensure complete reduction of HAuCl4. The solution turned deep red, indicating the formation of cit-Au NPs. For all the experiments (unless specified otherwise) a 2× diluted (with phosphate buffer) solution of the as-prepared cit-Au NP was used and the term cit-Au NP solution in the rest of the manuscript refers to this 2× diluted solution.Preparation of dinitrosalicylic acid (DNS) reagent
The reagent consisted of the following two components.Component ‘A’: 20.25 g of sodium potassium tartrate (Merck Specialists Private Limited, Mumbai, India) was dissolved in 60.0 mL MilliQ grade water to which 468.0 mg of phenol (Merck Specialists Private Limited, Mumbai, India) dissolved in 9.0 mL of 10% NaOH (Merck Limited, Mumbai, India) solution was added.
Component ‘B’: 300.0 mg of 3,5-dinitrosalicylic acid (SRL, Mumbai, India) was dissolved in 60.0 mL MilliQ grade water.
Preparation of enzyme solution and estimation of protein content in the 1.0 mg mL−1 enzyme solution
1.0 mg mL−1 of α-amylase (from hog pancreas, Fluka) solution was prepared by dissolving the required amount in phosphate buffer (pH = 7.0) followed by centrifugation at 5000 rpm to remove the insoluble part. The actual protein content in the final solution was calculated using the standard Bradford test35 for protein for 1.0 mg mL−1 enzyme solution and found to be equal to 0.07 mg mL−1. In the whole manuscript as well as in the supporting information the exact concentration of the enzyme solution, as estimated by the Bradford test, is mentioned. The test is usually carried out in the linear region of estimation using the Bradford method.36Enzyme activity by DNS method
75.0 μL of 0.07 mg mL−1 enzyme solution was added to 24.0 mL of 2× diluted cit-Au NPs (diluted from as-prepared solution with phosphate buffer) mixed well and left for 10 min. 2.0 mL of enz–Au NP composite thus obtained was added to various test tubes each containing 3.0 mL of 0.00, 1.67, 3.33, 5.00, 6.67, 8.33, 10.00, 11.67, 13.33, 15.00 and 16.67 mg mL−1 of starch solution (Merck Specialists Private Limited, Mumbai, India; prepared in phosphate buffer of pH 7.0) respectively. The final concentration of starch in each tube was thus equal to 0.0, 1.0, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0 and 10.0 mg mL−1 respectively and the enzyme concentration was 0.087 μg mL−1. All of these test tubes were then incubated in a water bath at 37 °C for 20 min. The reaction mixtures were then cooled down to room temperature. 1.0 mL of the reaction mixture was taken out from each test tube to which 1.0 mL each of ‘A’ and ‘B’ components of DNS reagent was added. All of these mixtures were then incubated at 95 °C for another 10 min, followed by cooling down to room temperature. 300.0 μL of the mixture thus obtained was added to 3.0 mL of Milli-Q water and the UV-vis spectrum was recorded using a Hitachi U-2900 spectrophotometer. The absorbance at 570 nm corresponding to each tube was converted to the respective maltose concentration by using the standard calibration curve obtained with maltose (refer to ESI Fig. S8†). The specific activity of the enzyme was then calculated using the following formula, Specific activity = amount of maltose produced/(volume of 1.0 mg mL−1 enzyme added × 10) where the factor 10 is the time period (in min) of incubation with the DNS reagent. However, the enzyme concentration was not 1.0 mg mL−1. Thus the volume of 1.0 mg mL−1 was calculated based on actual enzyme concentration (0.07 mg mL−1) and used for the calculation of specific activity. Similarly the specific activities with various concentrations of starch were calculated for 0.175 μg mL−1, 0.35 μg mL−1, 0.70 μg mL−1, 1.40 μg mL−1 and 2.80 μg mL−1 of final enzyme concentrations. For comparison control experiments (replacing cit-Au NPs with buffer) were carried out for each enzyme concentration. All the experiments were performed at least three times, where cit-Au NPs for each measurement was prepared separately and the average data of three sets have been reported.Calculation of Km, Vmax and kcat
For calculation of Km and Vmax, Lineweaver–Burk graphs were plotted for each enzyme concentration. The values of Km and Vmax were then calculated from the slope and the intercept values respectively. Additionally, kcat was calculated using the following relation:37kcat = μmoles of product/{μmoles of enzymes × time (in min)}
To check Au NP interference with the DNS test
Four different concentrations of maltose solution (2.50, 3.75, 5.00 and 7.50 mg mL−1 respectively) were prepared. To 3.0 mL of each solution, 2.0 mL of cit-Au NP was added so that the final concentrations of maltose in the solutions were 1.50, 2.25, 3.00 and 4.50 mg mL−1 respectively. 1.0 mL of the mixture was taken out from each test tube to which 1.0 mL each of ‘A’ and ‘B’ components of DNS reagent was added. The mixtures were then heated to 95 °C for 10 min and then cooled down to room temperature. 0.3 mL of the mixture thus obtained was added to 3.0 mL of Milli-Q water and the UV-vis spectrum was recorded using a Hitachi U-2900 spectrophotometer. Control experiments were performed for each of the maltose concentrations (by replacing cit-Au NPs with buffer). The absorbance at 570 nm was compared for each set (in the presence and absence of cit-Au NPs) to see the effect of cit-Au NPs, if any (refer to ESI Fig. S1†).Fluorescence of α-amylase in presence of Au NPs
12.5 μL of 0.07 mg mL−1 of α-amylase was added to 2.0 mL of 2× diluted cit-Au NP (i.e. equivalent to 0.175 μg mL−1 of enzyme concentration in the final reaction mixture of 5.0 mL), was mixed well and left for 10 min. The fluorescence spectrum of the enzyme both before and after addition of cit-Au NPs was recorded using a Horiba Jobin-yvon Fluoromax-4 spectrofluorometer, the excitation peak being set at 280 nm. Similarly fluorescence spectra were recorded for solutions containing 0.35 μg mL−1, 0.70 μg mL−1, 1.40 μg mL−1 and 2.80 μg mL−1 of enzyme concentration. For control studies, the fluorescence spectrum was recorded for each concentration of enzyme in buffer instead of in cit-Au NPs solution.Estimation of amount of free protein in solution
To 6.0 mL of 2× diluted cit-Au NPs solution 37.5 μL of 0.07 mg mL−1 of α-amylase was added, mixed well and the solution was left for 10 min. The ratio of enzyme:cit-Au NP was such that the final α-amylase concentration was equal to 0.175 μg mL−1 in the final reaction mixture in regular experiments. Similarly, four more sets were prepared with 75.0 μL, 150.0 μL, 300.0 μL and 600.0 μL of 0.07 mg mL−1 of α-amylase in 6.0 mL of cit-Au NP solution corresponding to 0.35 μg mL−1, 0.70 μg mL−1, 1.40 μg mL−1 and 2.80 μg mL−1 of α-amylase in regular experiments. The Au NP–enzyme (enz-Au NP) composites thus obtained were centrifuged at 20000 rpm in a Sigma 3–30 K high speed cold centrifuge for 20 min and the supernatants were collected. The supernatants were tested for the amount of protein by a simple method developed in our own laboratory using cit-Au NPs.29 Following this method, 0.1 mL to 0.4 mL of the supernatant solutions were added successively to 3.0 mL of cit-Au NP solution and the UV-vis spectrum was recorded after each addition using a Hitachi U-2900 spectrophotometer. The area under the curve (from 450 nm to 650 nm range) for each spectrum was calculated using the software in the instrument itself. The ratio of the area under the graph for each spectrum to that of the area under the graph for cit-Au NP only were then calculated after performing proper volume corrections to take care of the dilution factor. To exactly quantify the changes in area under the graphs, a calibration curve (ESI, Fig. S9B†) was prepared with the same stock of α-amylase and cit-Au NPs used. Using the values so obtained, the total amount of protein in the 6.0 mL supernatant could thus be calculated. It must be mentioned here that in each case a different dilution of the supernatant was used to obtain the best results. For example, the supernatants in Set 1 were used in the undiluted form, Set 2 was diluted two times, Set 3 and 4 were diluted five times and Set 5 was diluted to ten times. The results obtained were finally multiplied by the required factor to get the exact value. From the concentrations of the proteins in the supernatants the concentration of protein attached to the cit-Au NPs was obtained.
Preparation of TEM samples
A small part of the final reaction mixture containing 0.175 μg mL−1 of enzyme was taken after the reaction and drop cast onto a Cu grid and left to air-dry overnight. For preparation of the reaction mixture, refer to the previous section of the Experimental Section. Similarly, grids were prepared with reaction mixtures corresponding to 0.35 μg mL−1 and 2.80 μg mL−1 of enzyme respectively. These grids were further analyzed by a Jeol 2100 TEM machine (operated at a maximum voltage of 200 kV).Particle size analysis by DLS method
12.5 μL of 0.07 mg mL−1 of α-amylase was added to 2.0 mL of 2× diluted cit-Au NP to which 3.0 mL of buffer was added (which consists of 0.175 μg mL−1 of enzyme concentration), mixed well and was left for 10 min. 2.0 mL of this solution was taken in a cell for which particle size distribution was measured using Zetasizer Nano ZS (Malvern Instruments). Similarly, particle size distribution was measured for samples containing 0.35 μg mL−1 and 2.80 μg mL−1 of enzyme respectively.Modification of free thiol groups of α-amylase
In order to modify the free cysteine thiol groups of α-amylase, 100.0 μL of 6.0 mM 2,2′-dithiobis (5-nitropyridine) (DTNP) in dimethyl sulfoxide (DMSO) was added to 1.5 mL of 0.07 mg mL−1 enzyme solution, and the final reaction volume was made up to 9.5 mL with 100 mM phosphate buffer. The reaction mixture was stirred for 30 h at room temperature, which resulted in a yellow colored solution. The UV-vis spectrum of the sample was measured at 0 h and after 30 h of stirring to ascertain modification of the free cysteine thiol groups of α-amylase. The modified protein was then purified using dialysis upon which the yellow color of the solution disappeared. This dialyzed solution was used for further experiments.Acknowledgements
We thank the Department of Science and Technology (SR/S1/PC-30/2008), the Department of Biotechnology and Council of Scientific and Industrial Research, India for funds and a fellowship (to JD). We would also like to thank Dr Aiyagari Ramesh of the Department of Biotechnology, IIT Guwahati for his help and valuable suggestions, and Dr Eswaramoorthy and Mr Piyush Chaturbedy of JNCASR, Bangalore for their kind help with the DLS measurements.References
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120(9), 1959 CrossRef CAS.
- D. C. Hone, A. H. Haines and D. A. Russell, Langmuir, 2003, 19, 7141 CrossRef CAS.
- K. M. Mayer, S. Lee, H. Liao, B. C. Rostro, A. Fuentes, P. T. Scully, C. L. Nehl and J. H. Hafner, ACS Nano, 2008, 2(4), 687 CrossRef CAS.
- H. Wei, C. Chen, B. Han and E. Wang, Anal. Chem., 2008, 80(18), 7051 CrossRef CAS.
- J. Griffin and P. C. Ray, J. Phys. Chem. B, 2008, 112(36), 11198 CrossRef CAS.
- Z. Wang, R. Levy, D. G. Fernig and M. Brust, J. Am. Chem. Soc., 2006, 128, 2214 CrossRef CAS.
- Y. Choi, N-H. Ho and C-H. Tung, Angew. Chem., Int. Ed., 2007, 46, 707 CrossRef CAS.
- Y.-W. Jun, S. Sheikholeslami, D. R. Hostetter, C. Tajon, C. S. Craik and A. P. Alivisatos, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17735 CrossRef CAS.
- R. Bonomi, A. Cazzolaro, A. Sansone, P. Scrimin and L. J. Prins, Angew. Chem. Int. Ed., 2011, 50, 2307 CAS.
- T. Sawada, N. Shirakawa, Y. Hirai, M. Hashizume, M. Aizawa and T. Serizawa, Chem. Lett., 2011, 40, 142143 Search PubMed.
- J. Wang, L. Wu, J. Ren and X. Qu, Small, 2012, 8(2), 259 CrossRef CAS.
- S. Li, L. Mao, Y. Tian, J. Wang and N. Zhou, Analyst, 2012, 137, 823 RSC.
- P. Pandey, S. P. Singh, S. K. Arya, V. Gupta, M. Datta, S. Singh and B. D. Malhotra, Langmuir, 2007, 23, 3333 CrossRef CAS.
- G. K. Kouassi, J. Irudayaraj and G. McCarty, J. Nanobiotechnol., 2005, 3, 1 CrossRef.
- C. S. Wu, C. T. Wu, Y. S. Yang and F. H. Ko, Chem. Commun., 2008, 5327 RSC.
- C. C. You, S. S. Agasti, M. De, M. J. Knapp and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 14162 Search PubMed.
- B. J. Jordan, R. Hong, G. Han, S. Rana and V. M. Rotello, Nanotechnology, 2009, 20, 1 CrossRef.
- A. Dyal, K. Loos, M. Noto, S. W. Chang, C. Spagnoli, K. V. P. M. Shafi, A. Ulman, M. Cowman and R. A. Gross, J. Am. Chem. Soc., 2003, 125, 1684 CrossRef CAS.
- L. M. Rossi, A. D. Quach and Z. Rosenzweig, Anal. Bioanal. Chem., 2004, 380, 606 CrossRef CAS.
- B. J. Jordan, R. Hong, B. Gider, J. Hill, T. Emrick and V. M. Rotello, Soft Matter, 2006, 2, 558 RSC.
- G. K. Ahirwal and C. K. Mitra, Sensors, 2009, 9, 881 CrossRef CAS.
- J. L. Brennan, N. S. Hatzakis, T. R. Tshikhudo, N. Dirvianskyte, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan and M. Brust, Bioconjugate Chem., 2006, 17, 1373 CrossRef CAS.
- A. Rangnekar, T. K. Sarma, A. K. Singh, J. Deka, A. Ramesh and A. Chattopadhyay, Langmuir, 2007, 23(10), 5700 CrossRef CAS.
- A. Gole, C. Dash, V. Ramakrishnan, S. R. Sainkar, S. B. Mandale, M. Rao and M. Sastry, Langmuir, 2001, 17, 1674 CrossRef CAS.
- A. Verma, H. Nakade, J. M. Simard and V. M. Rotello, J. Am. Chem. Soc., 2004, 126, 10806 CrossRef CAS.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9(1), 308 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS.
- G. L. Miller, Anal. Chem., 1959, 31(3), 426 CrossRef CAS.
- J. Deka, A. Paul and A. Chattopadhyay, J. Phys. Chem. C, 2009, 113(17), 6936 CAS.
- C-S. Wu, C-C. Lee, C-T. Wu, Y-S. Yang and F-H. Ko, Chem. Commun., 2011, 47, 7446 RSC.
- J. L. Brennan, A. G. Kanaras, P. Nativo, T. R. Tshikhudo, C. Rees, L. C. Fernandez, N. Dirvianskyte, V. Razumas, M. Skjot, A. Svendsen, C. I. Jorgensen, R. Schweins, M. Zackrisson, T. Nylander, M. Brust and J. Barauskas, Langmuir, 2010, 26(16), 13590 CrossRef CAS.
- P. Sharma, S. Sathyanarayana, P. Kumar and K. C. Gupta, Anal. Biochem., 1990, 189, 173 CrossRef CAS.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, S. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(21), 8691 CrossRef CAS.
- D. Zhang, O. Neumann, H. Wang, V. M. Yuwono, A. Barhoumi, M. Perham, J. D. Hartgerink, P. Wittung-Stafshede and N. J. Halas, Nano Lett., 2009, 9(2), 666 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS.
- T. Zor and Z. Selinger, Anal. Biochem., 1996, 236, 302 CrossRef CAS.
- H. F. Gilbert, Basic concepts in biochemistry - A student’s survival guide, McGraw-Hill, United States of America, Search PubMed2nd edn, 2000.
Footnote |
| † Electronic Supplementary Information (ESI) available: contains the additional graphs and calculations involved and TEM images. See DOI: 10.1039/c2ra20056b/ |
| This journal is © The Royal Society of Chemistry 2012 |