Iridium catalyzed enantioselective hydrogenation of α-alkoxy and β-alkoxy vinyl ethers†
Ye
Zhu
and
Kevin
Burgess
*
Texas A&M University, Department of Chemistry, P.O. Box 30012, College Station, TX 77842-3012, USA. E-mail: burgess@tamu.edu; Fax: 979-845-8839; Tel: 979-845-4345
First published on 13th April 2012
Abstract
Iridium catalyzed, enantioselective hydrogenations of functionalized, acid-sensitive vinyl ethers are reported. These reactions are mediated by the chiral N-carbene–oxazoline complex 1 that, unlike similar N,P-ligated catalysts (6 and 7), gives hydrogenation products (with enantioselectivities up to 96%) without significant acid-mediated degradation. In general, higher enantioselectivities were observed for the allylic alcohol derivatives (2) than the α-methoxycinnamate-based compounds (4).
Introduction
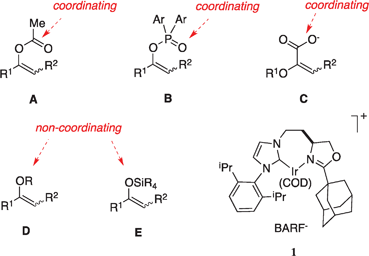
Asymmetric hydrogenations of vinyl ethers give the same outcome as enantioselective reductions of ketones.1–4 Unlike ketone reductions, however, they provide chiral ethers directly, and this can be advantageous in access to building blocks for the pharmaceutical and agrochemical industries.5–9 On the other hand, homogeneous hydrogenation reactions tend to produce protons10 and, since vinyl ethers are sensitive to acid, this limits application of the vinyl ether approach.Another limitation on hydrogenations of vinyl ethers relates to the fact that many of the most desirable substrates are hindered trisubstituted alkenes. Hindered alkenes without a proximal coordinating functional group (CFG) are not amenable to hydrogenations with catalysts we broadly refer to as Wilkinson's catalyst analogs (M(phosphine)2 catalysts where, for instance, M = Rh, Ir). Consequently, for vinyl ethers more complexes are likely to hydrogenate substrates A–C11–28 than for the functionalized, but not coordinatively functionalized, substrates D and E. Indeed, there are relatively few reports of asymmetric hydrogenation substrates in classes D and E, and the enantioselectivities observed have not reached levels that are practical for applications in organic syntheses (alkylated enol ethers,29–31 silylated enol ethers,32 furans31,33).
Crabtree's catalyst34 is the best known of an extremely small group of complexes that will mediate hydrogenation of hindered alkenes that do not contain coordinating functional groups (CFGs).35 Thus it would appear that chiral derivatives of Crabtree's catalyst might be ideal for asymmetric hydrogenations of substrates like D and E. Many chiral derivatives of Crabtree's catalyst with N,P-ligands are, in fact, not suitable because, as we have shown, protons are generated in these reactions and they tend to decompose the substrates before they can be hydrogenated. This assertion is consistent with work from the groups of Andersson36 and of Pfaltz37 for which they reported Ir–N,P catalysts are unsuitable for the more acid-sensitive substrates. Results from our laboratories10,38 indicated the carbene–oxazoline complex 139,40 is the most effective homogenous catalyst for asymmetric hydrogenations of substrates D–E reported to date. We have demonstrated that this is because 1 is less inclined to generate protons than similar N,P-ligated complexes,10 and it provides a stereoelectronic environment conducive to high enantioselectivities.41
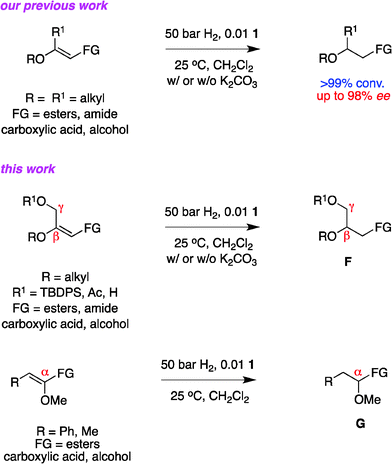
Studies for this paper were performed to elucidate the scope of catalyst 1 for asymmetric hydrogenations of other enol ethers without strongly coordinating functional groups, specifically the α,ω-functionalized chirons F and the α-oxygenated systems G. An attribute of building blocks F is that they can be homologated from either terminus. Attempted syntheses of chirons G provide opportunities to test how catalyst 1 would process a molecular alkene skeleton that is different to the β-oxygenated systems tested to date.
Results and discussion
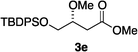
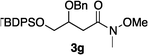
It would not be worthwhile to undertake the project outlined above unless there were convenient access to the enol ether substrates. Fortunately, there are good literature procedures for preparing β-alkoxy vinyl ethers 2d,e42via trimethylphosphine mediated conjugate additions of alcohols to electron-deficient alkynes (Scheme 1a). We developed these protocols using a deprotection/Weinreb amide43 formation/protection sequence to generate a broader range of substrates, 2a–c,f,g. Scheme 1b shows an aldol condensation route to substrates 4a–e.44 All these alkenes could be prepared with excellent E/Z selectivities (>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0) for the crude materials, and subsequent flash chromatographic purification afforded essentially pure E-vinyl ethers 2 and pure Z-vinyl ethers 4.
:1.0) for the crude materials, and subsequent flash chromatographic purification afforded essentially pure E-vinyl ethers 2 and pure Z-vinyl ethers 4.
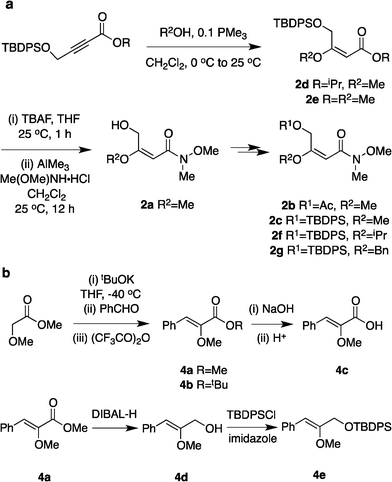 | ||
| Scheme 1 Syntheses of α-alkoxy and β-alkoxy vinyl ethers. | ||
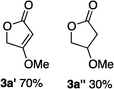
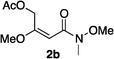
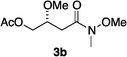
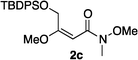
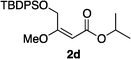
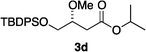
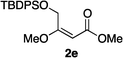
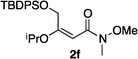
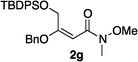
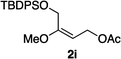
Hydrogenations of β-alkoxy vinyl ethers 2a–i were run using the same optimized conditions developed in our previous work (Table 1).38 High conversions could be achieved for all the vinyl ethers except for the sterically hindered alkene 2f, which cannot be hydrogenated at all under the same reaction conditions. The alkene 2a was highly labile to acid, and was converted to a cyclic vinyl ether completely during the hydrogenation. This is unsurprising since even our N,carbene–Ir catalyst generates protons during the hydrogenation (though less than the corresponding N,P-complexes).10 After 48 h, the cyclic unsaturated vinyl ether 3a′ was isolated as the major product and also crude 1H NMR identified about 30% hydrogenated product, saturated lactone 3a′′. Lactone 3a′′ presumably arose from the direct hydrogenation; we know this because the unsaturated lactone was not reduced under the same reaction conditions. Protection of the free side-chain hydroxy group in alkenes 2b and 2c circumvented the lactonization issue so the hydrogenation proceeded as expected. Use of a bulky protecting group afforded higher enantioselectivity in these reactions (cf2b (Ac), 74% ee; 2c (TBDPS), 94% ee). Size reduction of the carbonyl-substituent decreased the enantioselectivities (2c, 94% ee; 2d, 82% ee; 2e, 75% ee), but the highest enantioselectivity was obtained when the functional group was an alcohol, (2h, 96% ee). Only 64% ee was observed when that alcohol was converted to an acetate (alkene 2i), so a free-OH appears to be important for the high stereoselectivity. Hydrogenation of the β-benzyloxy vinyl ether 2g proceeded with high efficiency and enantioselectivity (>99% conv., 90% ee) providing a synthesis of the corresponding oxygenated chiral alcohol with an O-protecting group that we would expect to be removable via hydrogenolysis.
All the hydrogenation reactions reported in this paper went cleanly to product; even crude 1H NMR spectra were acceptable. In general, the crude products were passed through a silica plug (EtOAc/hexanes = 3:7) to get rid of catalyst, and then concentrated; the residue was concentrated to give essentially pure products. Consequently, the yields are in close agreement with the conversions observed.
|
|
|||
|---|---|---|---|
| 2 | Conv. (%)a | ee (%)b | 3 c |
| a Determined by 1H NMR. b Determined via analysis on a chiral GC column or chiral HPLC column. c Stereochemical assignments via comparison with authentic samples. d Assignment of the absolute configuration of the product was not made. e 1.0 equivalent of K2CO3 was added, the reaction time is 12 h. f The reaction time is 12 h. | |||
|
>99 | na |
|
|
>99 | 74 |
|
|
>99 | 94 |
|
|
>99 | 82 |
|
|
>99 | 75 |
|
|
na | na | na |
|
>99 | 90d |
|
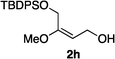
|
>99 | 96e |
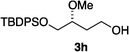
|
|
>99 | 64f |
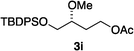
|
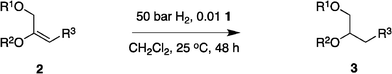
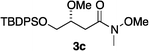
Our previous studies10 indicated N,P–Ir catalysts cannot hydrogenate the acid-sensitive enol ethers because they promote acid-mediated degradation. Here a simple experiment was performed to check the relative acidities of reaction solutions from the three different catalysts 1, 6 and 7. Proton NMR spectra of the crude hydrogenation products from these reactions show the N,carbene–Ir catalyst 1 gave almost exclusively the desired product 3c whereas the N,P–Ir catalyst 7 only gave a complex mixture (Fig. 1). The N,P-catalyst 6 gave a level of degradation that was between these extremes.
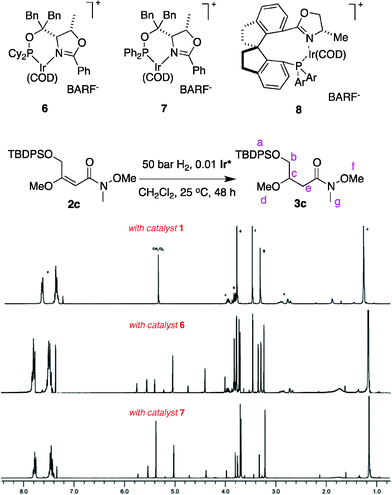 | ||
| Fig. 1 1H NMR spectra of the crude materials from the hydrogenation of 2c show no degradation with the N,carbene complex 1, more when the N,PCy2-system 6 was used, and the most degradation for the N,PPh2-catalyst 7. | ||
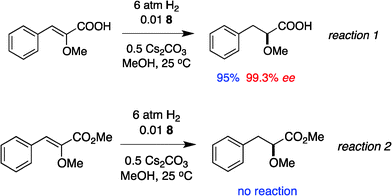
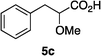
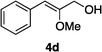
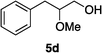
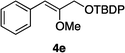
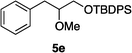
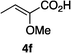
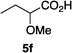
Optically enriched chiral α-alkoxy carboxylic acid derivatives are generally useful chirons.45 In 2010, Zhou's group reported their chiral spiro-N,P–iridium catalyst 8 mediates hydrogenation of α-methoxy cinnamic acid affording the corresponding chiral acid with 99% ee, but strong base was required (reaction 1).25 Others have used chiral diphosphine–Rh complexes at high temperature to obtain similarly good conversions and enantioselectivities.46 Zhou demonstrated that the corresponding ester, methyl α-methoxycinnamate was not hydrogenated under the same reaction conditions (reaction 2),25 so presumably the carboxylate was important in this reaction. Consequently, we decided to attempt to hydrogenate this same ester and others (4a–f) with catalyst 1. Table 2, shows that excellent conversions were achieved for all six alkenes, but the enantioselectivities were moderate, up to 61% (the allylic alcohol 4d).
|
|
||
|---|---|---|
| 4 | ee (%)a | 5 |
| a Determined via analysis on a chiral GC or HPLC column. | ||
|
5 |
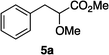
|
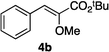
|
9 |
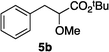
|
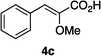
|
23 |
|
|
61 |
|
|
52 |
|
|
26 |
|
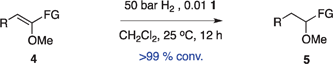
Conclusion
Chiral N,carbene–Ir catalyst 1 is effective for enantioselective hydrogenation of both α-alkoxy and β-alkoxy vinyl ethers. Hydrogenation of substrates 2 provides an easy access to α,γ-bifunctionalized chirons F, whereas similar N,P–Ir catalysts gave significant or even predominant degradation. The highest enantioselectivity in the series 2 compounds was for the alcohol 2h; our previous studies have indicated allylic alcohols tend not to be directly coordinating (whereas homoallylic alcohols are) so we prefer to describe the OH in this position as a FG rather than a CFG.41 Catalyst 1 also gives high conversions for α-methoxycinnamate substrates, but there is something about the shape of these substrates that is not conducive to high facial stereoselectivities.Experimental
General methods
NMR spectra were recorded in CDCl3. Flash chromatography was performed using silica gel (230–600 mesh) or basic Al2O3. Thin layer chromatography was performed using glass plates coated with silica gel 60 F254. All substrates were prepared under an atmosphere of nitrogen in oven-dried glassware with magnetic stirring. Air sensitive reagents and solutions were transferred via syringe and were introduced to the apparatus through rubber septa.General procedure for synthesis of vinyl ethers 2a–i
According to the method of Paintner and co-workers,42 a variety of alcohols was added to butynoate, using trimethylphosphine as a nucleophilic catalyst to give corresponding vinyl ether esters. Then a series of derivatives were made via regular methods.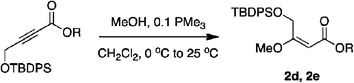
A solution of butynoate (5.0 mmol) in CH2Cl2 (20 mL) was added dropwise to a solution of methanol (15 mmol) and PMe3 (0.5 mL, 1.0 M in THF, 0.5 mmol) in CH2Cl2 (20 mL) at 0 °C by means of a syringe. Then the reaction mixture was allowed to warm to r.t. and stirred overnight. The solution was concentrated under reduced pressure. The crude product was purified by flash chromatography.
:70) gave the unsaturated lactone (0.43 g, 99%) as a white solid. The lactone was dissolved in CH2Cl2 (20 mL) and Me(OMe)NH·HCl (1.83 g, 18.8 mmol) was added in one portion followed by dropwise addition of AlMe3 solution (2 M in hexanes, 9.4 mL, 18.8 mmol). The resulting mixture was stirred at r.t. for 12 h. The reaction was then cooled to 0 °C, diluted with CH2Cl2 (20 mL) and H2O (5 mL) was carefully added. Saturated potassium sodium tartrate aqueous solution (20 mL) was then added. The resulting mixture was vigorously stirred for 1 h and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried (K2CO3) and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel, eluting with EtOAc/hexanes (50:50) gave the corresponding vinyl ether 2a (0.32 g, 49%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 5.68 (1H, s), 5.34 (1H, br), 4.31 (2H, s), 3.74 (3H, s), 3.69 (3H, s), 3.26 (3H, s); 13C NMR (75 MHz, CDCl3) δ 175.4, 169.1, 89.7, 63.5, 61.6, 55.9, 32.6. HRMS (ESI): calcd for C7H14NO4 [M + H]+ 176.0923. Found 176.0918.
:95) gave the desired product 2b (0.52 g, 100%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 5.71 (1H, s), 5.26 (2H, s), 3.72 (3H, s), 3.71 (3H, s), 3.23 (3H, s), 2.11 (3H, s); 13C NMR (75 MHz, CDCl3) δ 170.8, 167.5, 167.1, 91.5, 61.6, 56.0 (2 peaks), 32.6, 21.0. HRMS (ESI): calcd for C9H16NO5 [M + H]+ 218.1028. Found 218.1028.
:95) gave the corresponding vinyl ether 2c (0.47 g, 88%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.72–7.69 (4H, m), 7.49–7.34 (6H, m), 5.47 (1H, s), 4.96 (2H, s), 3.64 (3H, s), 3.63 (3H, s), 3.15 (3H, s), 1.08 (9H, s); 13C NMR (75 MHz, CDCl3) δ 171.4, 168.1, 135.9, 134.0, 129.7, 127.7, 89.2, 61.9, 61.5, 55.5, 32.6, 27.1, 19.7. HRMS (ESI): calcd for C23H32NO4Si [M + H]+ 414.2101. Found 414.2112.
:80) gave the desired product 2h (0.46 g, 76%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.74–7.70 (4H, m), 7.49–7.39 (6H, m), 4.85 (1H, t, J = 7.8 Hz), 4.26 (2H, s), 4.10–4.05 (2H, m), 3.53 (3H, s), 1.49 (1H, t, J = 5.7 Hz), 1.07 (9H, s); 13C NMR (75 MHz, CDCl3) δ 159.6, 135.9, 133.3, 130.0, 127.9, 99.4, 62.0, 58.4, 54.7, 27.0, 19.4. HRMS (ESI): calcd for C21H28KO3Si [M + K]+ 395.1445. Found 395.1437.
:95) gave the desired product 2i (0.55 g, 33%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.73–7.70 (4H, m), 7.45–7.38 (6H, m), 4.73–4.55 (3H, m), 4.27 (2H, s), 3.52 (3H, s), 2.02 (3H, s), 1.07 (9H, s); 13C NMR (75 MHz, CDCl3) δ 171.2, 160.7, 135.8, 133.5, 129.9, 127.8, 94.1, 61.6, 60.7, 54.7, 26.9, 21.3, 19.4. HRMS (ESI): calcd for C23H30NaO4Si [M + Na]+ 421.1811. Found 421.1806.
Synthesis of vinyl ethers 4a–f
Vinyl ethers 4c and 4f, are synthesized via the reported procedure.25 Hydrogenation products 5c and 5f, are known compounds. The characterization data for these compounds can be found in the reported literature and our characterization data agree well with those reported.47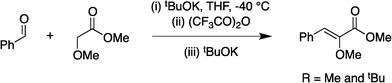
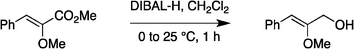 :80) gave the desired product 4d (0.68 g, 95%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.63–7.58 (2H, m), 7.36–7.28 (2H, m), 7.23–7.17 (1H, m), 5.64 (1H, s), 4.32 (2H, s), 3.85 (3H, s), 1.81 (1H, br); 13C NMR (75 MHz, CDCl3) δ 155.1, 135.8, 128.8, 128.5, 126.6, 109.6, 63.1, 56.3. HRMS (ESI): exact mass calcd for C10H13O2 [M + H]+ 165.0916. Found 165.0911.
:80) gave the desired product 4d (0.68 g, 95%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.63–7.58 (2H, m), 7.36–7.28 (2H, m), 7.23–7.17 (1H, m), 5.64 (1H, s), 4.32 (2H, s), 3.85 (3H, s), 1.81 (1H, br); 13C NMR (75 MHz, CDCl3) δ 155.1, 135.8, 128.8, 128.5, 126.6, 109.6, 63.1, 56.3. HRMS (ESI): exact mass calcd for C10H13O2 [M + H]+ 165.0916. Found 165.0911.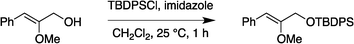 :95) gave the corresponding vinyl ether 4e (0.79 g, 98%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.82–7.16 (15H, m), 5.52 (1H, s), 4.34 (2H, s), 3.85 (3H, s), 1.11 (9H, s); 13C NMR (75 MHz, CDCl3) δ 154.8, 136.2, 135.9, 133.5, 130.1, 128.7, 128.4, 128.0, 126.2, 108.7, 63.8, 56.5, 27.0, 19.5. HRMS (ESI): exact mass calcd for C26H31O2Si [M + H]+ 403.2093. Found 403.2090.
:95) gave the corresponding vinyl ether 4e (0.79 g, 98%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.82–7.16 (15H, m), 5.52 (1H, s), 4.34 (2H, s), 3.85 (3H, s), 1.11 (9H, s); 13C NMR (75 MHz, CDCl3) δ 154.8, 136.2, 135.9, 133.5, 130.1, 128.7, 128.4, 128.0, 126.2, 108.7, 63.8, 56.5, 27.0, 19.5. HRMS (ESI): exact mass calcd for C26H31O2Si [M + H]+ 403.2093. Found 403.2090.
General catalytic hydrogenation conditions
The corresponding alkenes (0.1 mmol) were dissolved in CH2Cl2 (0.5 M) and (S)-1 (1 mol%) was then added. The resulting solution was degassed by three cycles of freeze–pump–thaw and then transferred to a Parr Bomb. The bomb was flushed with hydrogen for 1 min without stirring. The mixture was then stirred at 700 rpm under 50 bar H2. After 48 h (for esters and its derivatives) or 12 h (for alcohols and its derivatives), the bomb was vented and the solvent evaporated. The crude product was passed through a silica plug (EtOAc/hexanes = 3:7). The enantiomeric ratio of the crude material was then measured through chiral capillary GC analysis using a β- or γ-CD column48 (carrier gas: helium; column pressure: 18.21 psi; gas flow rate: 1.6 mL min−1; temperature: 135 °C) or through chiral HPLC analysis, which is performed with a 254 nm UV detector and a chiral column (Chiralcel OD).
Acknowledgements
Financial support for this work was provided by the National Science Foundation (CHE-0750193) and the Robert A. Welch Foundation (A1121).References
- R. Noyori and T. Ohkuma, Pure Appl. Chem., 1999, 71, 1493–1501 CrossRef CAS.
- R. Noyori, Acta Chem. Scand., 1996, 50, 380–390 CrossRef CAS.
- N. Menashe, E. Salant and Y. Shvo, J. Organomet. Chem., 1996, 514, 97–102 CrossRef CAS.
- D. G. Genov and D. J. Ager, Angew. Chem., Int. Ed., 2004, 43, 2816–2819 CrossRef CAS.
- S. Hanessian, Total Synthesis of Natural Products: The Chiron Approach, Pergamon: New York, 1983 Search PubMed.
- Y. J. Perron, W. F. Minor, C. T. Holdrege, W. J. Gottstein, J. C. Godfrey, L. B. Crast, R. B. Babel and L. C. Cheney, J. Am. Chem. Soc., 1960, 82, 3934–3938 CrossRef CAS.
- L. H. Mejorado, C. Hoarau and T. R. R. Pettus, Org. Lett., 2004, 6, 1535–1538 CrossRef CAS.
- G. M. Coppola and H. F. Schuster, alpha-Hydroxy Acids in Enantioselective Syntheses, John Wiley & Sons: New York, 1997 Search PubMed.
- H. U. Blaser and E. Schmidt, Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, Wiley-VCH: Weinheim, Germany, 2004 Search PubMed.
- Y. Zhu, Y. Fan and K. Burgess, J. Am. Chem. Soc., 2010, 132, 6249–6253 CrossRef CAS.
- M. J. Burk, J. Am. Chem. Soc., 1991, 113, 8518–8519 CrossRef CAS.
- Q. Jiang, D. Xiao, Z. Zhang, P. Cao and X. Zhang, Angew. Chem., Int. Ed., 1999, 38, 516–518 CrossRef CAS.
- M. J. Burk, Acc. Chem. Res., 2000, 33, 363–372 CrossRef CAS.
- N. W. Boaz, Tetrahedron Lett., 1998, 39, 5505–5508 CrossRef CAS.
- E. A. Broger, W. Burkart, M. Hennig, M. Scalone and R. Schmid, Tetrahedron: Asymmetry, 1998, 9, 4043–4054 CrossRef CAS.
- K. Iseki, Y. Kuroki, T. Nagai and Y. Kobayashi, Chem. Pharm. Bull., 1996, 44, 477–480 CrossRef CAS.
- H. M. Jung, J. H. Koh, M.-J. Kim and J. Park, Org. Lett., 2000, 2, 2487–2490 CrossRef CAS.
- Y. Chi and X. Zhang, Tetrahedron Lett., 2002, 43, 4849–4852 CrossRef CAS.
- W. Tang, D. Liu and X. Zhang, Org. Lett., 2003, 5, 205–207 CrossRef CAS.
- M. T. Reetz, L. J. Goossen, A. Meiswinkel, J. Paetzold and J. F. Jensen, Org. Lett., 2003, 5, 3099–3101 CrossRef CAS.
- S. Wu, W. Wang, W. Tang, M. Lin and X. Zhang, Org. Lett., 2002, 4, 4495–4497 CrossRef CAS.
- L. Qiu, J. Wu, S. Chan, T. T.-L. Au-Yeung, J.-X. Ji, R. Guo, C.-C. Pai, Z. Zhou, X. Li, Q.-H. Fan and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5815–5820 CrossRef CAS.
- P. Cheruku, S. Gohil and P. G. Andersson, Org. Lett., 2007, 9, 1659–1661 CrossRef CAS.
- P. Cheruku, J. Diesen and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 5595–5599 CrossRef CAS.
- S. Li, S.-F. Zhu, J.-H. Xie, S. Song, C.-M. Zhang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 1172–1179 CrossRef CAS.
- P. E. Maligres, S. W. Krska and G. R. Humphrey, Org. Lett., 2004, 6, 3147–3150 CrossRef CAS.
- X. Cheng, J.-H. Xie, S. Li and Q.-L. Zhou, Adv. Synth. Catal., 2006, 348, 1271–1276 CrossRef CAS.
- W. Chen, P. J. McCormack, K. Mohammed, W. Mbafor, S. M. Roberts and J. Whittall, Angew. Chem., Int. Ed., 2007, 46, 4141–4144 CrossRef CAS.
- H. Leopold, S. Hardo and B. Helga, Angew. Chem. Int. Ed., 1968, 12, 942 Search PubMed.
- T. Hayashi, M. Tanaka and I. Ogata, Tetrahedron Lett., 1977, 295–296 CrossRef CAS.
- T. Ohta, T. Miyake, N. Seido, H. Kumobayashi and H. Takaya, J. Org. Chem., 1995, 60, 357–363 CrossRef CAS.
- T. Tanaka, Y. Watanabe, T. Mitsudo, Y. Yasunori and Y. Takegami, Chem. Lett., 1974, 137–140 CrossRef.
- S. Kaiser, S. P. Smidt and A. Pfaltz, Angew. Chem., Int. Ed., 2006, 45, 5194–5197 CrossRef.
- R. H. Crabtree, H. Felkin and G. E. Morris, J. Organomet. Chem., 1977, 141, 205–215 CrossRef CAS.
- X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS.
- P. Cheruku, J. Diesen and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 5595–5599 CrossRef CAS.
- N. S. Goulioukina, T. Y. M. Dolgina, G. N. Bondarenko, I. P. Beletskaya, M. M. Ilyin, V. A. Davankov and A. Pfaltz, Tetrahedron: Asymmetry, 2003, 14, 1397–1401 CrossRef CAS.
- Y. Zhu and K. Burgess, Adv. Synth. Catal., 2008, 350, 979–983 CrossRef CAS.
- M. T. Powell, D.-R. Hou, M. C. Perry, X. Cui and K. Burgess, J. Am. Chem. Soc., 2001, 123, 8878–8879 CrossRef CAS.
- M. C. Perry, X. Cui, M. T. Powell, D.-R. Hou, J. H. Reibenspies and K. Burgess, J. Am. Chem. Soc., 2003, 125, 113–123 CrossRef CAS.
- Y. Fan, X. Cui, K. Burgess and M. B. Hall, J. Am. Chem. Soc., 2004, 126, 16688–16689 CrossRef CAS.
- F. F. Paintner, M. Metz and G. Bauschke, Synthesis, 2002, 869–874 CrossRef CAS.
- J. L. Levin, E. Turos and S. M. Weinreb, Synth. Commun., 1982, 12, 989–993 CrossRef CAS.
- I. N. Houpis, L. E. Patterson, C. A. Alt, J. R. Rizzo, T. Y. Zhang and M. Haurez, Org. Lett., 2005, 7, 1947–1950 CrossRef CAS.
- G. M. Coppola and H. F. Schuster, alpha-Hydroxy Acids in Enantioselective Syntheses, VCH, 1997 Search PubMed.
- W. Chen, P. J. McCormack, K. Mohammed, W. Mbafor, S. M. Roberts and J. Whittall, Angew. Chem., Int. Ed., 2007, 46, 4141–4144 CrossRef CAS.
- J. J. Verendel, T. Zhou, J.-Q. Li, A. Paptchikhine, O. Lebedev and P. G. Andersson, J. Am. Chem. Soc., 2010, 132, 8880–8881 CrossRef CAS.
- D. U. Staerk, A. Shitangkoon and G. Vigh, J. Chromatogr., A, 1995, 702, 251–257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for the compounds 4a–f and 5a–f are reported. 1H and 13C NMR spectra of compounds. See DOI: 10.1039/c2ra01350a |
| This journal is © The Royal Society of Chemistry 2012 |