Reversible polymerization of novel monomers bearing furan and plant oil moieties: a double click exploitation of renewable resources
Carla
Vilela
a,
Letizia
Cruciani
a,
Armando J. D.
Silvestre
a and
Alessandro
Gandini
*ab
aCICECO and Chemistry Department, University of Aveiro, Campus de Santiago, 3810-193, Aveiro, Portugal
bMaterials Engineering Department, Engineering School of São Carlos, University of São Paulo, 13566-590, São Carlos, Brazil. E-mail: agandini@iqsc.usp.br
First published on 25th January 2012
Abstract
Monomers based on plant oil derivatives bearing furan heterocycles appended through thiol-ene click chemistry were prepared and, subsequently, polymerized via a second type of click reaction, i.e. the Diels–Alder (DA) polycondensation between furan and maleimide complementary moieties. Two basic approaches were considered for these DA polymerizations, namely (i) the use of monomers with two terminal furan rings in conjunction with bismaleimides (AA + BB systems) and (ii) the use of a protected AB monomer incorporating both furan and maleimide end groups. This study clearly showed that both strategies were successful, albeit with different outcomes, in terms of the nature of the ensuing products. The application of the retro-DA reaction to these polymers confirmed their thermoreversible character, i.e. the clean-cut return to their respective starting monomers, opening the way to original macromolecular materials with interesting applications, like mendability and recyclability.
Introduction
Polymers from renewable resources have acquired a respectable status within the macromolecular community and their scope is widening incessantly.1 Among the numerous sources of novel materials, plant oils occupy a prominent position, further enhanced by the growing research activities of the last several years,2–10 focussed in particular on the application of metathesis, thiol-ene chemistry and other original mechanistic approaches. The resulting materials include polyurethanes,11 polyesters,12 and other macromolecular structures2–9 in which the long aliphatic segments play a critical role in imparting properties like hydrophobicity and low thermal transitions, whereas the associated polar moieties favour biodegradability.Our interest in plant oils stems from the idea of applying previous knowledge on the use of furan monomers and furan chemistry to these substrates, through the joint exploitation of two click-chemistry mechanisms, viz. the thiol-ene and the Diels–Alder (DA) reactions.
The thiol-ene addition, known for over 100 years, is a well documented reaction,13–16 which has attracted the interest of researchers due to its click-chemistry character.17 It has been successfully used for the synthesis of star polymers,18 dendrimers19 and disaccharides,20 among others. In the field of oleochemistry, the thiol-ene coupling with plant oils or their fatty acids has been applied to prepare several monomers and polymers.21–27 For instance, Samuelsson et al.21 and Claudino et al.22 investigated the kinetics of the photo-initiated thiol-ene coupling of trifunctional thiols with methyl oleate and methyl linoleate. Bantchev et al.23 prepared sulphide-modified plant oils through the thiol-ene addition of butanethiol to canola and corn oils for such end-use applications as lubricants. Moreover, Türünç and Meier25 prepared a set of novel monomers derived from 10-undecenoic acid via thiol-ene additions in the absence of solvent and initiator, and their subsequent polymerization yielded a family of linear and hyperbranched polyesters with good thermal properties.
The Diels–Alder (DA) reaction, known since 1928, is another prominent example of click chemistry28 that has been studied very extensively in a variety of contexts,29–31 including notably the realm of furan polymers because of its potential in the preparation of various macromolecular architectures. These materials, based on renewable resources, possess, in addition to this important feature, promising properties in terms of thermoreversibility, mendability and recyclability.32–35 The important peculiarity of the DA reaction, apart from its click connotation, is its reversible character, illustrated here (Scheme 1) in the case of the coupling of a furan ring (diene) with a maleimide complementary moiety (dienophile), where the temperature is a key factor in determining the position of the equilibrium, which can be shifted heavily from predominant adduct formation (DA reaction), up to ca. 65 °C, to the predominant reversion to its precursors (retro-DA reaction), above ca. 100 °C.32–38 The kinetic characteristics associated with the course of the forward and backward reactions depend on the specific structure of the substituents attached to both heterocycles, and, of course, the reactant concentration, the medium and the temperature. The application of the DA reaction to polymer syntheses based on furan/maleimide reversible couplings has gained much attention in the last few decades. Two different approaches dominate this realm, i.e. (i) polycondensation reactions calling upon complementary bifunctional or polyfunctional monomers, including AnBm-type structures, and (ii) reversible cross-linking of linear polymers bearing pendant furan or maleimide moieties, based on the temperature sensitivity of the DA equilibrium.32,33 The fact that the forward DA reaction gives rise to both endo and exo stereoisomer adducts39 does not play a significant role in these macromolecular syntheses, since both participate in the chain growth.
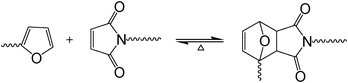 | ||
| Scheme 1 The DA equilibrium between growing species bearing, respectively, furan and maleimide end-groups. | ||
The present study, a follow-up to a preliminary communication that set the stage for this approach,40 describes the use of undecenyl compounds as suitable substrates for appending terminal DA functions, viz either two furan (A) heterocycles or a combination of a furan (A) and a protected maleimide (B) end-group. This was achieved by calling upon the thiol-ene reaction in conjunction with more classical chemical condensations. The ensuing AA and protected AB monomers were then polymerized through the DA polycondensation, the former with a bismaleimide (BB), the latter on its own, after liberating the maleimide moiety through the retro-DA reaction of the inert adduct.
Results and discussion
Synthesis and characterization of AA, AA′ and protected AB monomers
As far as we know, the thiol-ene coupling of 2-furylmethanethiol (FT) with alkenyl functions has never been reported before our preliminary communication40 and it was therefore vital to investigate it first with simple model compounds. The reactivity of FT towards both terminal and internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturations was assessed under various conditions using compounds bearing a single alkene moiety. The occurrence of the thiol-ene click reaction was confirmed using vinyl isobutyl ether and vinyl stearate (i.e. compounds with terminal unsaturations), with which coupling did take place, although at more modest rates than with conventional aliphatic thiols like 1,3-propanedithiol. The use of reagents incorporating internal CHCH groups, like methyl oleate (MO), resulted in a very sluggish process and it was therefore decided to concentrate our study on reagents bearing end unsaturations. The possibility of a retarding effect associated with the steric hindrance induced by the furan ring was excluded, since the aromatic counterpart, Ph–CH2–SH, did not display any appreciable rate decrease compared with the aliphatic homologues.
C unsaturations was assessed under various conditions using compounds bearing a single alkene moiety. The occurrence of the thiol-ene click reaction was confirmed using vinyl isobutyl ether and vinyl stearate (i.e. compounds with terminal unsaturations), with which coupling did take place, although at more modest rates than with conventional aliphatic thiols like 1,3-propanedithiol. The use of reagents incorporating internal CHCH groups, like methyl oleate (MO), resulted in a very sluggish process and it was therefore decided to concentrate our study on reagents bearing end unsaturations. The possibility of a retarding effect associated with the steric hindrance induced by the furan ring was excluded, since the aromatic counterpart, Ph–CH2–SH, did not display any appreciable rate decrease compared with the aliphatic homologues.
As mentioned above, the basic structure chosen as a typical and viable plant oil derivative was ω-undecenoic acid (UDA), i.e. the prominent fragment arising from the pyrolysis of castor oil,41 together with its reduced derivative undec-10-en-1-ol (UDOL). Two approaches were adopted to append furan end-groups onto UDA (Scheme 2), viz. (i) esterification with furfuryl alcohol (FA) at the carboxylic terminal and ene reaction with FT at the unsaturated counterpart, or (ii) esterification with allyl alcohol, followed by a double ene reaction with FT at both alkenyl end-groups. In both cases, α,ω-difuran monomers (AA and AA′) were obtained and characterized. The corresponding alcohol UDOL was used to prepare the protected AB monomer (Scheme 2) through esterification of its primary OH end-group with the protected 4-maleimidobutyric acid (MBA) followed by the ene reaction of its terminal unsaturation with FT. The protection of its maleimide group in the form of its furan-DA adduct, prevented the premature DA polycondensation of the intrinsically reactive furan/maleimide moieties during its synthesis, purification and storage.37,38
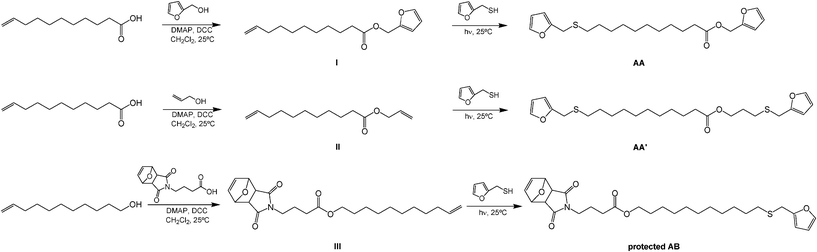 | ||
| Scheme 2 Synthetic pathways for the AA, AA′ and protected AB monomers. | ||
Both the bis-dienes AA and AA′ and the protected AB monomer were characterized by FTIR, Raman, 1H and 13C NMR spectroscopy.
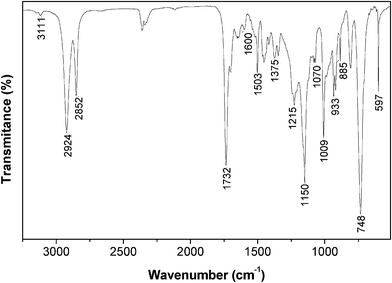
The FTIR spectrum of the AA monomer (Fig. 1) clearly showed the presence of furan heterocycle bands at 3111, 1600, 1503, 1375, 1215, 1070, 1009, 933, 885, 748, 597 cm−1,42 and of the ester CO and C–O bands at 1735 and 1150 cm−1, respectively.43 The Raman spectrum clearly indicated the presence of the C–S stretching vibration at 607 cm−1 and the absence of the SH band of the thiol moiety in the range 2590–2550 cm−1.43 The 1H NMR spectrum confirmed the expected structure through the appearance of typical resonances of the methylene protons of the ester moiety at δ 5.1 ppm (OCH2–2-furan), the methylene protons attached to the S atom at δ 2.5 (SCH2(CH2)n) and 3.7 ppm (SCH2–2-furan), and the furan heterocycle protons at about δ 6.2 (3-H), 6.3 (4-H), 7.3 (5-H) ppm for 2-furan–CH2S and δ 6.4 (4-H), 6.5 (3-H), 7.4 (5-H) ppm for 2-furan–CH2O, apart from the typical resonances of the methylene protons of UDA's aliphatic chain at δ 1.3 (–(CH2)n–), 1.6 (CH2CH2C(O)) and 2.3 (CH2C(O)) ppm. The 13C NMR spectrum, apart from the unchanged resonances related to the aliphatic chain carbons (δ 24.8, 28.3–29.3 and 34.1 ppm), showed resonances at δ 31.7 (CH2SCH2–2-furan), 57.8 (OCH2–2-furan), 107.2 and 110.4 (furan C-4 and C-3), 142.0 (C-5 of 2-furan–CH2S), 143.2 (C-5 of 2-furan–CH2O), 149.6 (C-2 of 2-furan–CH2S), 151.9 (C-2 of 2-furan–CH2O) and the COester peak at 173.5 ppm.
Similarly, the changes in the FTIR, Raman, 1H and 13C NMR spectra associated with the functionalization of UDA to give the AA′ monomer (Scheme 2) displayed the same features, confirming that the esterification and the thiol-ene reactions had indeed taken place equally well, as given in detail in the Experimental section.
The FTIR spectrum of the protected AB monomer was consistent with the structure shown in Scheme 2, by the presence of all the relevant peaks, viz. (i) the asymmetric and symmetric CO stretching vibrations of the imide group at 1772 and 1696 cm−1, respectively,44 (ii) the furan heterocycle bands at 3110, 1602, 1503, 1399, 1245, 1160, 1069, 1009, 917, 878, 735 and 598 cm−1,42 and (iii) the CO ester band at 1731 cm−1, together with the absence of the OH band of primary alcohols and the OH and CO bands of the carboxylic group.43 Although this technique did not succeed in detecting the weak ν (C–S) signal, its identification was possible by Raman spectroscopy with a band at 606 cm−1.43 The 1H NMR spectrum clearly confirmed this structure through the presence of the protons of the protected maleimide moiety at about 2.9 (CHCHCHCO), 5.3 (CHCHCHCO) and 6.5 (CHCHCHCO) ppm and the methylene protons of the ester group at δ 4.1 ppm (OCH2), as well as of the 2-furan ring protons at δ 6.2 (3-H), 6.3 (4-H) and 7.4 (5-H) ppm and the methylene protons attached to the S atom at δ 2.5 (SCH2(CH2)n) and 3.7 ppm (SCH2–2-furan). The 13C NMR spectrum of the protected AB monomer was also in tune with the proposed structure with, in addition to the carbon resonances related to the aliphatic chain (δ 25.8, 28.5 and 28.8–29.4 ppm), the furan ring carbons (δ 107.2, 110.3, 142.0 and 151.9 ppm) and the protected maleimide carbon resonances (δ 47.3, 81.0, 136.5 and 176.2 ppm), it was also possible to observe signals resonating at δ 31.7 and 64.7 ppm, which were readily identified as the methylenic carbons of the thioether moiety (SCH2) and of the ester group (OCH2), respectively.
The common structural feature of these three monomers, besides their terminal DA-reactive furan or maleimide functions, was the long methylene sequence, i.e. the flexible bridge joining them. This suggests that all the ensuing DA polymers, namely those derived from AA and AA′ with the equally flexible bridge joining the aliphatic bismaleimide (BMH), and that formed by the self-polycondensation of the deprotected AB monomer, were expected to have relatively low glass transition temperatures.
Diels–Alder polymers
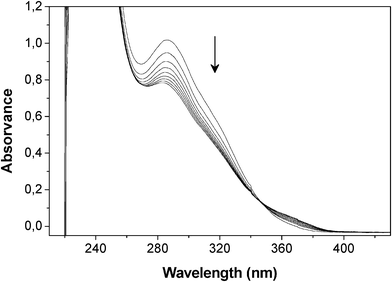
Given that no published information could be found on the reactivity of FT as a diene in its DA interaction with maleimide dienophiles, preliminary experiments were carried out on model compounds. Furfuryl methyl sulphide (FMS) was found to react with N-methylmaleimide (MM) to form the expected adduct, as followed by UV spectroscopy (Fig. 2), and the same occurred with the larger sulphide arising from the ene reaction of FT with vinyl stearate. Additionally, the DA polymerization of furfuryl sulphide (FS) with 1,6-bismaleimidohexane (BMH) took place, as expected, to give the corresponding linear polyadduct (Scheme 3) with an Mw of 5500 and a Tg of −11 °C. All these experiments were carried out in both UV cells and NMR tubes and the course of the corresponding DA reactions followed, respectively, by the progressive decrease in (i) the maleimide peak at 300 nm (loss of the OC–CC–CO conjugation, as shown in the example of Fig. 2) and (ii) the resonance intensity of the furan and maleimide protons, associated with the formation of the DA adducts. Compared with other systems previously studied in our laboratory,36,37 all these reactions, conducted in the same conditions of concentration, medium and temperature, were found to proceed more slowly, suggesting that the 2-Fu–CH2–S– group played a (modest) retarding role. Despite this minor quantitative difference, the qualitatively positive outcome of these tests opened the way to the study of the programmed polymerization systems.
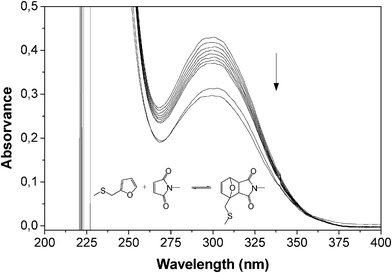 | ||
| Fig. 2 Evolution of the UV spectrum during the DA reaction between FMS (0.1 M) and MM (0.1 M) in TCE at 65 °C during 7 h (spectra taken hourly) and then at 25 °C after 3 and 7 days. | ||
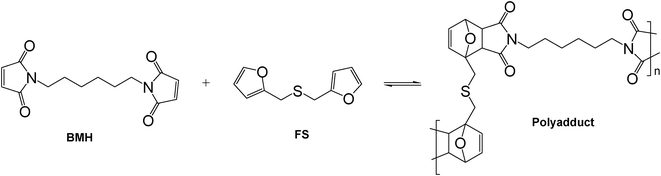 | ||
| Scheme 3 Model Diels–Alder polycondensation between BMH and FS. | ||
The complementary bis-dienophile selected as the DA-polycondensation comonomer for both the AA and AA′ bis-dienes was 1,6-bismaleimidohexane (BMH), i.e. a monomer bearing, as mentioned above, an equally flexible aliphatic bridging structure, leading to the formation of polymers P1 and P2, respectively, as exemplified in Scheme 4 for P2. All polymerizations were followed by both UV and 1H NMR spectroscopy.
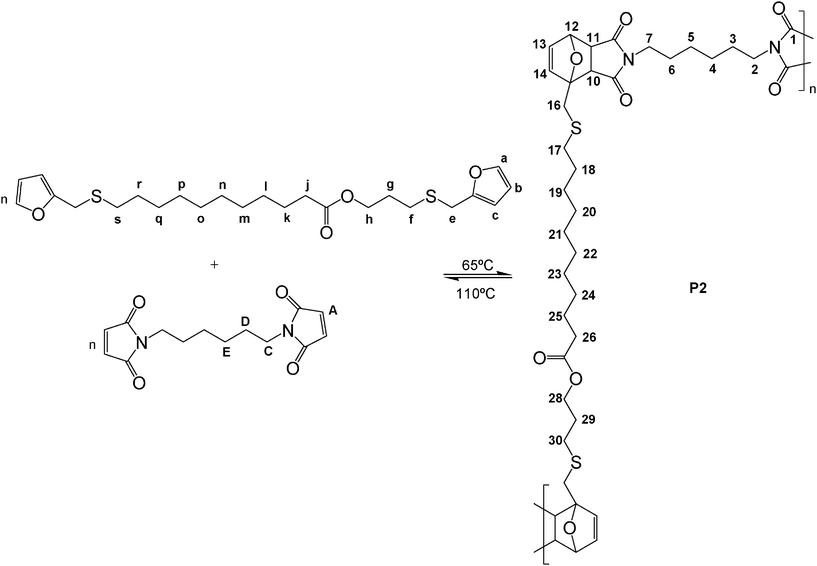 | ||
| Scheme 4 DA polycondensation between the difunctional AA′ and BMH monomers. | ||
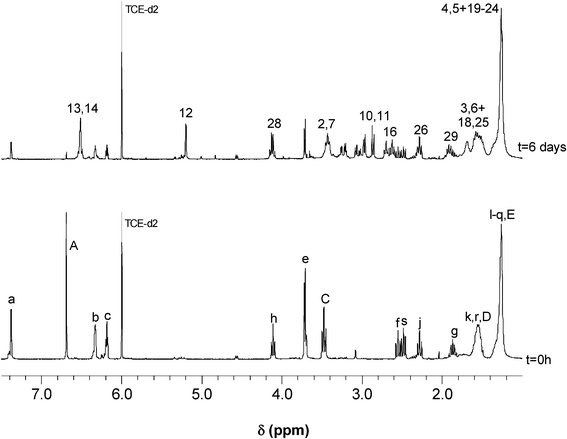
The DA polymerization of AA and AA′ at 65 °C, followed by UV spectroscopy for 8 h, revealed a progressive decrease in the optical density of the maleimide peak at about 300 nm, as shown in Fig. 3 for P1, accompanied by a corresponding increase in the viscosity of the medium. As reported in previous studies,36–38 the spectral pattern gave rise to an isosbestic point (progressive replacement of the maleimide peak at ∼300 nm by the absorption of the unconjugated carbonyl groups of the adduct at ∼260 nm), which suggested the occurrence of a single reaction pathway, viz. the DA condensation. The concomitant changes in the 1H NMR spectra (as illustrated for P2 in Fig. 4) displayed a gradual decrease in the intensity of the maleimide (δ 6.7 ppm, CHCH) and furan (c–H at δ 6.2 ppm, b–H at δ 6.3 ppm and a–H at δ 7.4 ppm) proton resonances at a rate similar to that of the UV spectra evolution and the simultaneous surge of the peaks associated with the three sets of protons assigned to the polymer adducts, at about δ 2.9 (10-H and 11-H), 5.3 (12-H) and 6.5 ppm (13-H and 14-H).
 | ||
| Fig. 3 Evolution of the UV spectrum of the maleimide peak during the DA reaction between AA (0.1 M) and BMH (0.1 M) at 65 °C in TCE for 8 h. | ||
 | ||
| Fig. 4 Evolution of 1H NMR spectrum of the DA polymerization between AA′ and BMH at 65 °C for 6 days (see Scheme 4 for peak assignment). | ||
Once these polymerizing systems had reached high conversion, both polymers were then submitted to the corresponding retro-DA depolymerisation at 110 °C, followed by 1H NMR spectroscopy. These processes were characterized by the reverse pattern with respect to the polycondensations, consisting in the gradual decrease in the adduct resonance (δ 2.9, 5.3 and 6.5 ppm) intensities and the corresponding growth of the furan (δ 6.2, 6.3 and 7.4 ppm) and maleimide (δ 6.7 ppm) counterparts, together with the decrease in viscosity of the solutions. Within a few days, the spectra revealed the presence of resonances of the starting monomers (Fig. 5 (b)), thus confirming the complete thermo-reversible nature of these systems. The solutions of the regenerated monomers were allowed to cool to 65 °C and a second polymerization took place (Fig. 5 (c)), emphasizing the reproducibility of these cyclic events, as previously reported.36–38
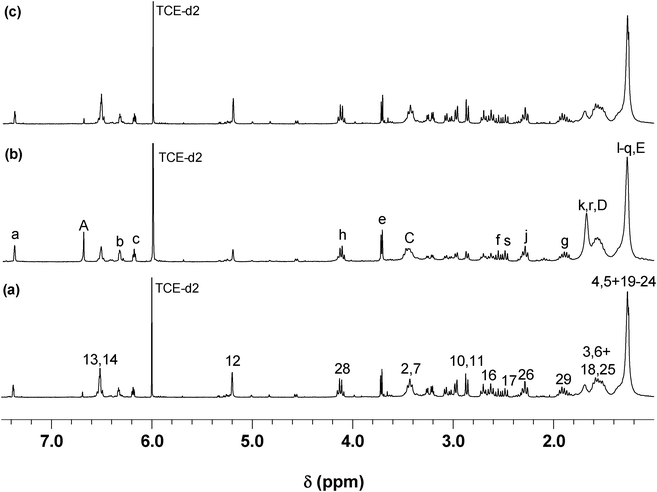 | ||
| Fig. 5 1H NMR spectrum of (a) P2, (b) retro-DA of P2 after 3 days, and (c) second DA polymerization after 6 days (see Scheme 4 for peak assignment). | ||
The use of an AB monomer bearing both the furan and the maleimide moieties in its structure represents an interesting alternative for linear DA polymerizations, as already shown,37,38 since it provides the ideal initial stoichiometry. The synthesis of the AB monomer generated a molecule with a protected maleimide moiety in order to avoid premature polymerization. This stable furan-DA adduct became ready for polymerization after the in situ deprotection of the masked end group by the retro-DA reaction at 110 °C, which released furan and regenerated the maleimide moiety. The polycondensation could then be initiated by decreasing the temperature to 65 °C. The 1H NMR spectrum of the ensuing AB monomer, despite the evidence of incipient polymerization, clearly confirmed its total deprotection through the presence of the free maleimide protons at δ 6.7 ppm (CHCH), plus the absence of the adduct peaks at 2.9 (CHCHCHCO), 5.3 (CHCHCHCO) and 6.5 (CHCHCHCO) ppm.
The evolution of the DA polymerization of the deprotected AB monomer, leading to the formation of P3, was also followed by UV and 1H NMR spectroscopy and more qualitatively by the regular increase in the viscosity of the reaction medium. The progress of the UV spectrum with reaction time mimicked the features exemplified in Fig. 3, exhibiting again a characteristic isosbestic point. The 1H NMR monitoring of the polymerizing system revealed all the details of the progressive decrease in the signals ascribed to the unreacted furan (δ 6.2, 6.3 and 7.4 ppm) and maleimide moieties (δ 6.7 ppm) and the corresponding increase of those associated with the polyadduct protons at δ 6.5 (CHCHCHCO), 5.3 (CHCHCHCO) and 2.9 (CHCHCHCO) ppm. After the system had attained high conversion, the retro-DA depolymerisation was followed at 110 °C for three days by 1H NMR spectroscopy. The expected behaviour was once more observed, viz. the increase in the peaks of the furan and maleimide protons to the detriment of the adduct signals, as previously discussed. Thereafter, the system was brought back to 65 °C and left for a number of days in order to promote a second polymerization. The 1H NMR spectrum of the ensuing polymer was analogous to that taken at the end of the first polymerization, thus confirming the reversible nature of this polymerizing system.
The isolated P1, P2 and P3 polymers in the form of sticky materials were characterized by DSC and SEC. Table 1 shows their glass transition temperature (Tg), molecular weight distribution, weight-average degree of polymerization (DPw) and polydispersity index (PDI). The DSC analyses were stopped at 80 °C, due to the prevalence of the retro-DA reaction above this temperature. As anticipated, the Tg of these polymers was well below room temperature as a result of the flexible nature of the spacing moieties separating the adducts. The much higher molecular weight of P3 confirmed the advantage of using an AB monomer, with its intrinsically unitary A/B molar ratio. The SEC tracings showed evidence of the presence of cyclic oligomers, including the corresponding dimers, through the appearance of individual peaks within the distribution curves, as depicted in Fig. 6 for P1 and P2. Given the relatively low monomer concentrations used in these DA polycondensations, the occurrence of cyclization seems reasonable, as indeed already observed in a recent study on the DA polymerization of other AB monomers.38
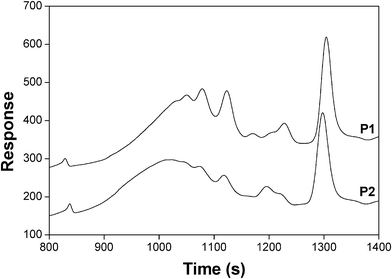 | ||
| Fig. 6 SEC tracing of P1 and P2. | ||
| Polymer | Tg °C | Mw | Mn | DPw | PDI | |
|---|---|---|---|---|---|---|
| P1: | AA + BMH | −40 | 6500 | 4500 | 10 | 1.4 |
| P2: | AA′ + BMH | −28 | 9100 | 5800 | 13 | 1.5 |
| P3: | AB | −2 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
9000 | 37 | 1.8 |
It is important to emphasize that the fact that these polymerizations proceeded at a rather slow pace also stems from the modest monomer concentrations adopted here in order to allow UV and NMR spectra to be conveniently monitored. In practical terms, the same systems could obviously be made to polymerise more rapidly at higher concentrations, including in bulk. Another relevant aspect to be underlined here is that no attempt was made to optimize the other conditions for the DA polymerizations, so that the molecular weights measured for the corresponding materials do not reflect the highest achievable values.
Experimental
Materials
Allyl alcohol (Aldrich, ≥ 99%), benzyl mercaptan (BM, Aldrich, 99%), 1,6-bismaleimidohexane (BMH, Tyger Scientific Inc.), chloroform-d (CDCl3, Acros Organics, 99.8 atom% D), N,N′-dicyclohexylcarbodiimide (DCC, Aldrich, 99%), 4-dimethylaminopyridine (DMAP, Sigma), furan (Acros Organics, 99%), furfuryl alcohol (FA, Aldrich, 98%), furfuryl methyl sulfide (FMS, SAFC, ≥ 97%), furfuryl sulfide (FS, Aldrich, ≥ 98%), 2-furylmethanethiol (FT, Aldrich, 98%), N-methylmaleimide (Aldrich, 97%), 4-maleimidobutyric acid (MBA, Fluka, ≥ 98%), methyl oleate (MO, Aldrich, 99%), phenylethyl mercaptan (SAFC, ≥ 98%), 1,3-propanedithiol (Aldrich, 99%), 1,1,2,2-tetrachloroethane (TCE, Sigma-Aldrich, ≥ 98.0%), 1,1,2,2-tetrachloroethane-d2 (TCE-d2, Acros Organics, 99.5 atom% D), 10-undecenoic acid (UDA, Aldrich, 98%), 10-undecen-1-ol (UDOL, Aldrich, 98%), vinyl isobutyl ether (Aldrich, 99%) and vinyl stearate (Aldrich, 95%) were used as received without any further purification. Irgacure 651 (2,2-dimethoxy-2-phenylacetophenone, DMPA) was generously supplied by BASF-The Chemical Company, Ludvigshafen. Polystyrene standards with molecular weights between 1700 and 66000 were supplied by Polymer Laboratories. Other chemicals and solvents were of laboratory grade.
Characterization methods
The FT-IR spectra were taken with a Perkin Elmer FT-IR System Spectrum BX Spectrometer equipped with a single horizontal Golden Gate ATR cell. Each spectrum was an average of 32 scans taken with 4 cm−1 resolution in the 500–4000 cm−1 range.Raman spectra were measured after 3500 scans with 4 cm−1 resolution using a Bruker RFS 100/S FTRaman spectrometer (Nd: YAG laser, 1064 nm excitation) at a power of 150 mW. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 NMR spectrometer operating at 300 and 75 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm), relative to the internal standard tetramethylsilane (TMS, δ = 0.00 ppm).
Electronic spectra were taken with a temperature-controlled Jasco V-560 spectrophotometer using 1 cm Hellma Suprasil cells equipped with 9.9 mm quartz spacer and a quartz-to-pyrex graded seal.
DSC thermograms were obtained with a Perkin Elmer Diamond DSC unit using aluminium pans under nitrogen with a heating rate of 20 °C min−1 in the temperature range −90 to 80 °C.
The molecular weights and molecular weight distributions of the polymers were determined by size-exclusion chromatography (SEC) with a Varian PL-GPC 110 instrument equipped with an IR-PD 2020 light scattering detector, using N,N′-dimethylacetamide (DMA) as the mobile phase, a run time of 30 min and a column temperature of 70 °C. Polystyrene standards were used for narrow standard calibration.
Synthesis of 4-(3,6-epoxy-1,2,3,6-tetrahydrophthalimido)butanoic acid (protected MBA)
4-Maleidobutyric acid (MBA) was used in the synthesis of the AB-type monomer below, thus its maleimide group was previously protected in order to avoid premature DA condensation of the maleimide and furan moieties. A solution of MBA (5 mmol) and furan (7.5 mmol) in TCE (5 mL) was prepared and left for 72 h at 65 °C. The remaining furan and the solvent were removed under vacuum. The ensuing pallid yellow solid was then dissolved in dichloromethane, reprecipitated in an excess of diethyl ether, filtered and dried under vacuum. The final product was obtained in 95% yield. δH (300 MHz, TCE-d2, Me4Si): 6.5 (2H, s,CHCHCHCO), 5.3 (2H, s, CHCHCHCO), 3.6 (2H, t, J = 6.8 Hz, NCH2CH2), 2.9 (2H, s, CHCHCHCO), 2.3 (2H, t, J = 7.4 Hz, CH2C(O)), 1.9 (2H, m, NCH2CH2). δC (75 MHz, TCE-d2, Me4Si): 177.6 (COacid), 176.3 (COmaleimide), 136.4 (CHCHCHCO), 80.9 (CHCHCHCO), 47.3 (CHCHCHCO), 37.9 (NCH2CH2), 30.8 (CH2C(O)), 22.5 (NCH2CH2).
Direct esterification reactions
The esterification of the UDA and protected MBA carboxylic acids, in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP), was adapted from the method reported by Rauf and Parveen,45 in which the direct use of DCC as an activating agent promotes esterification under very mild conditions. The selected carboxylic acid (4.0 mmol), DCC (4.0 mmol) and DMAP (0.40 mmol) were added to a solution of the given alcohol (4.0 mmol) in dichloromethane (20 ml). The reaction mixture was left overnight at room temperature under nitrogen and thereafter the precipitated N,N′-dicyclohexylurea (DCU), formed during the reaction, was filtered off and the filtrate washed with water (2 × 20 mL), 5% acetic acid solution (2 × 20 mL) and again water (2 × 20 mL). The additional DCU was again removed by filtration and the organic phase dried over anhydrous sodium sulphate before vacuum removal of the solvent. The desired compounds were obtained as viscous liquids in excellent yields (≥ 90%).CH), 5.1 (2H, s, OCH2), 5.0 and 4.9 (2H, m, CH2CH), 2.3 (2H, t, J = 7.5 Hz, CH2C(O)), 2.0 (2H, m, CH2CHCH2), 1.6 (2H, m, CH2CH2C(O)), 1.3 (10H, m, aliphatic CH2). δC (75 MHz, CDCl3, Me4Si): 173.4 (CO), 149.5 (C-2 of 2-furan), 143.1 (C-5 of 2-furan), 139.1 (CH2CH), 114.1 (CH2CH), 110.4 (C-3 of 2-furan), 106.5 (C-4 of 2-furan), 57.8 (OCH2–2-furan), 34.1 (CH2–C(O)), 33.7 (CH2CHCH2), 28.8-29.2 (aliphatic CH2), 24.8 (CH2CH2C(O)).
CH2), 5.8 (1H, m, CH2CH(CH2)n), 5.3 and 5.2 (2H, m, OCH2CHCH2), 5.0 and 4.9 (2H, m, CH2CH(CH2)n), 4.6 (2H, dt, J = 5.8, 1.4 Hz, OCH2), 2.3 (2H, t, J = 7.5 Hz, CH2C(O)), 2.0 (2H, m, CH2CHCH2(CH2)n), 1.6 (2H, m, CH2CH2C(O)), 1.3 (10H, m, aliphatic CH2). δC (75 MHz, CDCl3, Me4Si): 173.4 (CO), 139.0 (CH2CH(CH2)n), 132.3 (OCH2CHCH2), 118.0 (OCH2CHCH2), 114.1 (CH2CH(CH2)n), 64.8 (OCH2CHCH2), 34.2 (CH2C(O)), 33.7 (CH2CHCH2(CH2)n), 29.2–28.8 (aliphatic CH2), 24.8 (CH2CH2C(O)).
CHCHCHCO), 5.8 (1H, m, CH2CH(CH2)n), 5.3 (2H, s, CHCHCHCO), 5.0 and 4.9 (2H, m, CH2CH(CH2)n), 4.1 (2H, t, J = 6.8 Hz, OCH2), 3.6 (2H, t, J = 6.9 Hz, NCH2CH2), 2.9 (2H, s, CHCHCHCO), 2.3 (2H, t, J = 7.5 Hz, CH2C(O)), 2.0 (2H, m, CH2CHCH2(CH2)n), 1.9 (2H, m, NCH2CH2), 1.6 (2H, m, OCH2CH2), 1.3 (12H, m, aliphatic CH2). δC (75 MHz, CDCl3, Me4Si): 176.1 (COmaleimide), 172.7 (COester), 139.2 (CH2CH(CH2)n), 136.4 (CHCHCHCO), 114.1 (CH2CH(CH2)n), 80.8 (CHCHCHCO), 64.7 (OCH2(CH2)n), 47.2 (CHCHCHCO), 37.9 (NCH2CH2), 33.7 (CH2CHCH2(CH2)n), 31.1 (CH2C(O)), 29.3–28.8 (aliphatic CH2), 28.4 (OCH2CH2), 25.8 (OCH2CH2CH2), 22.7 (NCH2CH2).
Thiol-ene click reactions
In a typical reaction, the ene (3 mmol), 2-furylmethanethiol (6 mmol) and Irgacure 651 (0.06 mmol) were placed in a glass flask and thoroughly degassed with a stream of nitrogen to remove the air, since the thiol is air sensitive. The reaction mixture was irradiated for 4 h at room temperature using a UV-light source (Vilber Lourmat UV lamp VL-6-LC, 230V∼50/60 Hz) working at 365 nm. The excess thiol was then vacuum removed and the ensuing monomers were obtained in near-quantitative yields as viscous liquids.O), 1600, 1503 and 1375 (furan ring stretching), 1215 and 1070 (furan in-plane CH deformation), 1150 (C–O stretching), 1009 (ring breathing), 933, 885 and 748 (furan C–H out-of-plane deformation), 607 (C–S), 597 (ring deformation). δH (300 MHz, CDCl3, Me4Si): 7.4 (1H, dd, J = 2.0, 0.7 Hz, 5-H of 2-furan–CH2O), 7.3 (1H, dd, J = 1.6, 0.7 Hz, 5-H of 2-furan–CH2S), 6.5 (1H, d, J = 3.0 Hz, 3-H of 2-furan–CH2O), 6.4 (1H, dd, J = 3.2, 1.8 Hz, 4-H of 2-furan–CH2O), 6.3 (1H, dd, J = 3.1, 1.9 Hz, 4-H of 2-furan–CH2S), 6.2 (1H, d, J = 3.5 Hz, 3-H of 2-furan–CH2S), 5.1 (2H, s, OCH2–2-furan), 3.7 (2H, s, SCH2–2-furan), 2.5 (2H, t, J = 7.4 Hz, CH2SCH2–2-furan), 2.3 (2H, t, J = 7.5 Hz, CH2C(O)), 1.6 (2H, m, CH2CH2C(O)), 1.5 (2H, m, CH2CH2S), 1.3 (12H, m, aliphatic CH2). δC (75 MHz, CDCl3, Me4Si): 173.5 (CO), 151.9 (C-2 of 2-furan–CH2O), 149.6 (C-2 of 2-furan–CH2S), 143.2 (C-5 of 2-furan–CH2O), 142.0 (C-5 of 2-furan–CH2S), 110.4 (2-furan C-3), 107.2 (2-furan C-4), 57.8 (OCH2-2-furan), 34.1 (CH2C(O)), 31.7 (CH2S), 28.3–29.3 (aliphatic CH2), 24.8 (CH2CH2C(O)). Its EI-MS spectrum, obtained by GC-MS, gave a molecular ion at m/z 378, consistent with its C21H30O4S molecular formula.
O stretching), 1593, 1502 and 1383 (furan ring stretching), 1244, 1167 and 1070 (furan in-plane CH deformation), 1149 (C–O stretching), 1008 (ring breathing), 933, 885 and 732 (furan C–H out-of-plane deformation), 608 (C–S), 598 (ring deformation). δH (300 MHz, CDCl3, Me4Si): 7.4 (2H, dd, J = 1.7, 0.8 Hz, 2-furan 5-H), 6.3 (2H, dd, J = 3.1, 1.8 Hz, 2-furan 4-H), 6.2 (2H, d, J = 3.3 Hz, 2-furan 3-H), 4.2 (2H, t, J = 6.3 Hz, OCH2), 3.7 (4H, s, SCH2–2-furan), 2.6 (2H, t, J = 7.5 Hz, OCH2CH2CH2S), 2.5 (2H, t, J = 7.5 Hz, SCH2CH2(CH2)n), 2.3 (2H, t, J = 7.5 Hz, CH2C(O)), 1.9 (2H, m, OCH2CH2CH2S), 1.6 (2H, m, CH2CH2C(O)), 1.5 (2H, m, SCH2CH2(CH2)n), 1.3 (12H, m, aliphatic CH2). δC (75 MHz, CDCl3, Me4Si): 173.8 (CO), 151.8 (2-furan C-2), 142.0 (2-furan C-5), 110.3 (2-furan C-4), 107.3 (2-furan C-3), 62.8 (OCH2), 34.3 (CH2C(O)), 31.7 (SCH2), 28.1–29.3 (aliphatic CH2), 24.9 (CH2CH2C(O)). The GC-MS molecular ion at m/z 452 was in tune with its C24H36O4S2 molecular formula.
O stretching), 1602, 1503 and 1399 (furan ring stretching), 1245, 1160 and 1069 (furan in-plane CH deformation), 1150 (C–O stretching), 1009 (ring breathing), 917, 878 and 735 (furan C–H out-of-plane deformation), 606 (C–S), 598 (ring deformation). δH (300 MHz, CDCl3, Me4Si): 7.4 (1H, dd, J = 1.7, 0.7 Hz, 2-furan 5-H), 6.5 (2H, s, CHCHCHCO), 6.3 (1H, dd, J = 3.1, 1.9 Hz, 2-furan 4-H), 6.2 (1H, d, J = 3.4 Hz, 2-furan 3-H), 5.3 (2H, s, CHCHCHCO), 4.1 (2H, t, J = 6.7 Hz, OCH2), 3.7 (2H, s, SCH2–2-furan), 3.6 (2H, t, J = 6.6 Hz, NCH2CH2), 2.9 (2H, s, CHCHCHCO), 2.5 (2H, t, J = 7.5 Hz, SCH2CH2(CH2)n), 2.3 (2H, t, J = 7.5 Hz, CH2C(O)), 1.9 (2H, m, NCH2CH2), 1.6 (2H, m, OCH2CH2), 1.5 (2H, m, SCH2CH2(CH2)n), 1.3 (14H, m, aliphatic CH2). δC (75 MHz, CDCl3, Me4Si): 176.2 (COmaleimide), 172.7 (COester), 151.9 (2-furan C-2), 142.0 (2-furan C-5), 136.5 (CHCHCHCO), 110.3 (2-furan C-4), 107.2 (2-furan C-3), 81.0 (CHCHCHCO), 64.7 (OCH2), 47.3 (CHCHCHCO), 38.1 (NCH2CH2), 31.7 (CH2S), 31.2 (CH2C(O)), 29.4–28.8 (aliphatic CH2), 28.5 (OCH2CH2), 25.8 (OCH2CH2CH2), 22.8 (NCH2CH2). The GC-based data gave a molecular ion at m/z 518 that matched its C28H39NO6S molecular formula.
Diels–Alder reactions
AA + BB—A typical procedure applied to the use of UV spectroscopy follows: 0.1 M solutions of monomer AA (18.9 mg, 0.05 mmol) or AA′ (22.6 mg, 0.05 mmol) and BMH (13.8 mg, 0.05 mmol) in TCE (0.5 mL) were prepared separately and poured in a 0.1 mm Suprasil cell, which was then placed in the temperature-controlled spectrophotometer at 65 °C, where UV spectra were taken at regular intervals.A typical procedure applied to the use of 1H NMR spectroscopy follows: stoichiometric quantities (0.1 mmol) of the two monomers (AA or AA′ and BMH) were dissolved in 1.0 mL of TCE-d2, introduced in an NMR tube and the initial 1H NMR spectrum taken at room temperature. The tube was then kept in an oil bath at 65 °C and spectra taken at regular intervals. The ensuing polymers were isolated by precipitation into a large excess of 40–60 °C petroleum ether, followed by filtration, dissolution in dichloromethane and solvent removal under reduced pressure.
A fraction of the ensuing polymers was dissolved in TCE or TCE-d2 (1.0 mL) and the retro-DA depolymerisation reaction followed at 110 °C by UV and 1H NMR spectroscopy.
AB —The protected monomer (0.1 mmol) was dissolved in 1 mL of TCE-d2 and the solution brought to 110 °C while a gentle stream of nitrogen was bubbled through it for several hours to remove the furan generated by the retro-DA of the maleimide adduct. The unprotected AB monomer was then allowed to polymerize at 65 °C and the reaction followed by UV and 1H NMR spectroscopy. The ensuing polymer was precipitated in an excess of 40–60 °C petroleum ether, filtered, dissolved in dichloromethane and vacuum dried. Again, the depolymerisation of the DA polymer was carried out at 110 °C.
Conclusion
The successful application of the thiol-ene coupling to append furan moieties to molecules derived from plant oils was the first positive outcome of this investigation. Its second relevant contribution has to do with the exploitation of the Diels–Alder polycondensation of the novel monomers for the preparation of functional macromolecular materials based on renewable resources with original properties and promising applications, like mendability, recyclability and controlled phase changes.Work is in progress to further the insight into these systems and to develop other monomers based on furans and plant oils.
Acknowledgements
The authors wish to thank the Portuguese Foundation for Science and Technology (FCT) for analytical instrumentation support (POCI 2010 and REEQ/515/CTM/2005) and for a doctorate grant to Carla Vilela (SFRH/BD/44884/2008).References
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- N. M. Belgacem and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. N. M. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008; ch. 3, pp 39–66 Search PubMed.
- V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2008, 33, 1199–1215 CrossRef CAS.
- Z. S. Petrovič, Polym. Rev., 2008, 48, 109–155 CrossRef.
- Y. Lu and R. C. Larock, ChemSusChem, 2009, 2, 136–147 CrossRef CAS.
- J. O. Metzger, Eur. J. Lipid Sci. Technol., 2009, 111, 865–876 CrossRef CAS.
- M. Galià, L. Montero de Espinosa, J. C. Ronda, G. Lligadas and V. Cádiz, Eur. J. Lipid Sci. Technol., 2010, 112, 87–96 CrossRef.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Biomacromolecules, 2010, 11, 2825–2835 CrossRef CAS.
- H. Miyagawa, A. K. Mohanty, R. Burgueño, L. T. Drzal and M. Misra, Ind. Eng. Chem. Res., 2006, 45, 1014–1018 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- M. Fiore, A. Marra and A. Dondoni, J. Org. Chem., 2009, 74, 4422–4425 CrossRef CAS.
- J. Samuelsson, M. Jonsson, T. Brinck and M. Johansson, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6346–6352 CrossRef CAS.
- M. Claudino, M. Johansson and M. Jonsson, Eur. Polym. J., 2010, 46, 2321–2332 CrossRef CAS.
- G. B. Bantchev, J. A. Kenar, G. Biresaw and M. G. Han, J. Agric. Food Chem., 2009, 57, 1282–1290 CrossRef CAS.
- C. Lluch, J. C. Ronda, M. Galià, G. Lligadas and V. Cádiz, Biomacromolecules, 2010, 11, 1646–1653 CrossRef CAS.
- O. Türünç and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 1822–1826 CrossRef.
- O. Türünç and M. A. R. Meier, Green Chem., 2011, 13, 314–320 RSC.
- C. Lluch, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Macromol. Rapid Commun., 2011, 32, 1343–1351 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- T. J. Brocksom, J. Nakamura, M. L. Ferreira and U. Brocksom, J. Braz. Chem. Soc., 2001, 12, 597–622 CrossRef CAS.
- F. Fringuelli and A. Taticchi, The Diels–Alder Reaction: Selected Practical Methods, John Wiley & Sons, West Sussex, 2002 Search PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- A. Gandini and M. N. Belgacem, ACS Symp. Ser., 2007, 954, 280–295 CrossRef CAS.
- A. Gandini, Polym. Chem., 2010, 1, 245–251 RSC.
- A. Gandini, in Green Polymerisation Methods: Renewable Starting Materials, Catalysis and Waste Reduction, ed. R. T. Mathers and M. A. R. Meier, Wiley-VCH, Weinheim, 2011, ch. 3, pp. 29–53 Search PubMed.
- A. Gandini, in Biopolymers: New Materials for Sustainable Films and Coatings, ed. D. Plackett, John Wiley & Sons, Weinheim, 2011, ch. 9, pp. 179–209 Search PubMed.
- A. Gandini, D. Coelho and A. J. D. Silvestre, Eur. Polym. J., 2008, 44, 4029–4036 CrossRef CAS.
- A. Gandini, D. Coelho and A. J. D. Silvestre, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2053–2056 CrossRef CAS.
- A. Gandini, D. Coelho and A. J. D. Silvestre, Polym. Chem., 2011, 2, 1713–1719 RSC.
- H. Kwart and I. Burchuk, J. Am. Chem. Soc., 1952, 74, 3094–3097 CrossRef CAS.
- C. Vilela, L. Cruciani, A. J. D. Silvestre and A. Gandini, Macromol. Rapid Commun., 2011, 32, 1319–1323 CrossRef CAS.
- M. Van der Steen and C. V. Stevens, ChemSusChem, 2009, 2, 692–713 CrossRef CAS.
- A. R. Katritzky and J. M. Lagowski, J. Chem. Soc., 1959, 657–660 RSC.
- L. J. Bellamy, The Infra-red Spectra of Complex Molecules, Chapman and Hall, London, 3rd edn, 1975, vol. 1 Search PubMed.
- H. K. Hall and R. Zbinden, J. Am. Chem. Soc., 1958, 80, 6428–6432 CrossRef CAS.
- A. Rauf and H. Parveen, Eur. J. Lipid Sci. Technol., 2004, 106, 97–100 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |