Synthetic 5′-phosphorylated oligodeoxynucleotide purification through catching full-length sequences by polymerization†
Yinan
Yuan
ab,
Suntara
Fueangfung
a,
Xi
Lin
a,
Durga
Pokharel
a and
Shiyue
Fang
*a
aDepartment of Chemistry, Michigan Technological University, 1400 Townsend Drive, Houghton, Michigan 49931, USA. E-mail: shifang@mtu.edu; Fax: +1 (906)487-2061; Tel: +1 (906)487-2023
bSchool of Forest Resources and Environmental Science, Michigan Technological University, 1400 Townsend Drive, Houghton, Michigan 49931, USA
First published on 5th January 2012
Abstract
The readily scalable catching by polymerization purification technology has been further advanced to purify 5′-phosphorylated synthetic oligodeoxynucleotides (ODNs). The new technology utilizes a phosphoramidite that contains a fluoride-cleavable diisopropylsilyl acetal linker and a polymerizable methacrylamide group, and is capable of phosphorylation of ODN. For purification, the phosphoramidite was coupled to the 5′-end of full-length ODN on a synthesizer. Because failure sequences were capped in each synthetic cycle, only the full-length sequences were phosphinylated and acrylated. After cleavage and deprotection, the crude ODN was subjected to polymerization under typical acrylamide gel formation conditions. The full-length ODN was incorporated into polymer. The failure sequences and other impurities were simply removed by washing with water. Pure full-length ODN that contained a 5′-phosphate group was cleaved from the polymer with HF–pyridine. Reversed-phase (RP) HPLC showed that the ODN was pure, and the recovery yield was higher than that of typical preparative HPLC purification.
Introduction
Synthetic oligonucleotides (ONs) including ODNs and oligoribonucleotides and their unnatural analogs have found wide applications in molecular biology and other scientific areas.1–8 These ONs are synthesized on an automated synthesizer using protected nucleoside phosphoramidite monomers.9 Typically, the first monomer is anchored to a solid support via its 3′-OH group through a cleavable linker. Phosphoramidite monomers are coupled to the 5′-OH in a stepwise fashion. Each synthetic cycle consists of detritylation, coupling, capping and oxidation steps, which finishes the addition of one nucleoside. Excess reagents and side products are washed away after each step while the nascent ON remains on the solid support. Normally, the coupling step is highly efficient, but generation of truncated failure sequences is unavoidable. This problem is more serious for long ONs (more than 50-mer) and large-scale ON syntheses.10 To ease purification, the failure sequences are capped with reagents such as acetic anhydride so that they do not participate in reactions in the subsequent synthetic cycles. After synthesis, the ONs are cleaved from the solid support and fully deprotected. The crude ON, which mainly contains full-length ON, failure sequences and small organic molecule impurities resulting from deprotection, is then purified with various methods. The small organic molecules are easy to remove because they have very different physical properties from ONs. However, the separation of full-length sequence from the failure ones is difficult due to their identical physical properties. Currently used methods for ON purification include gel electrophoresis, RP HPLC, anion exchange HPLC, RP cartridge extraction, fluorous affinity extraction,10–11 biotin–avidin enabled affinity purification,12–14 and other techniques.15 All these methods have limitations as we discussed in a previous paper.16 Briefly, gel electrophoresis can only be used to purify minute quantities of ON. HPLC methods need expensive instrument, consume large volumes of buffer, are labor intensive, and are expensive to scale up. RP cartridge extraction gives less pure ON. Fluorous affinity extraction and biotin–avidin enabled affinity purification require solid phase extraction materials, which are normally not reusable. To solve the ON purification problem, we recently developed two methods that utilized the concept of catching by polymerization for ODN purification.16–17 In one method, the failure sequences were capped with an acrylated phosphoramidite in each synthetic cycle. After synthesis, purification was achieved by simply polymerizing the failure sequences.17 In the second method, the full-length sequences were coupled to an acrylated phosphoramidite. After synthesis, purification was achieved by polymerization of full-length sequences, washing away the impurities and cleaving pure ODN from polymer.16 In this paper, we report our results on using the catching by polymerization technology to purify synthetic ODNs that contain a 5′-phosphate group.Results and discussion
In some biological applications such as ligation of synthetic ODNs for gene synthesis,18 cloning,19 mutagenesis,20 and ligation chain reaction,21 a phosphate group at the 5′-end of ODN is required. This group can be installed via enzyme-catalyzed phosphorylation.19 A more convenient approach is to attach the group on an automated DNA synthesizer. Several reagents have been commercialized for the purpose.22–23 One example is the phosphoramidite of 4,4′-dimethoxytrityl (DMTr)-protected bis(hydroxymethyl)malonate.23 The DMTr group in this reagent serves as a hydrophobic handle for RP HPLC purification after synthesis. At the end of purification, the group is removed and the phosphate group at the 5′-end of ODN is released under basic conditions. In this paper, we combine this chemical phosphorylation technology with our newly developed catching by polymerization ODN purification technology to develop a method for purification of 5′-phosphorylated ODN.The reagent we designed for this ODN phosphorylation and purification application is phosphoramidite 1. Its synthesis is shown in Scheme 1. The easily accessible and known methacryloyl tertiary alcohol 216 was coupled to diethyl bis(hydroxymethyl)malonate with dichlorodiisopropylsilane to give 3. The alcohol 3 was then phosphinylated to give the target phosphoramidite 1. To demonstrate the usefulness of the phosphorylation and purification technique, the acrylated ODN 4 (see Scheme 2) was synthesized on a DNA synthesizer. Typical UltraMild ODN synthesis conditions, which use the more base-labile phenoxyacetyl protecting groups and allow deprotection with concentrated ammonium hydroxide at room temperature (see experimental section for details), were used. At the end of the synthesis, the phosphoramidite 1 was coupled to the 5′-end of the ODN for 5 min. Oxidation and capping in this cycle were carried out under normal conditions. The cleavage and deprotection were carried out on the synthesizer. The crude ODN contained the full-length sequence 4, and impurities, which included the failure sequences 5 and small molecules from protecting groups. Because the failure sequences were capped with phenoxyacetic anhydride (Pac2O) in each synthetic cycle, they did not react with the phosphoramidite 1, and did not contain a methacrylamide group. Only the full-length sequences were acrylated. The crude ODN was analyzed with RP HPLC. As shown in Fig. 1, trace a, the failure sequences 5 appeared at around 19 min. Due to the relatively more hydrophobic tag at the 5′-end of 4, the full-length ODN had an unusually long retention time of 62 min. The small peaks appearing at 31 and 59 min may be attributed to small molecules from protecting groups.
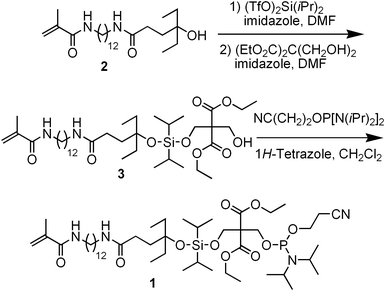 | ||
| Scheme 1 Synthesis of acrylation and phosphinylation phosphoramidite 1. | ||
 | ||
| Fig. 1 RP HPLC traces of ODNs: (a) crude 4; (b) failure sequences 5; (c) purified 8; (d) crude 61-mer 9; (e) purified 61-mer 10; (f) crude 25-mer 11 synthesized using normal protecting groups; (g) purified 25-mer 12. | ||
The crude ODN was subjected to polymerization under typical polyacrylamide gel formation conditions (Scheme 2). Specifically, N,N-dimethylacrylamide was used as the monomer, a small amount of N,N′-methylenebis(acrylamide) was used as the cross-linker, water was used as the solvent, and the polymerization was conveniently initiated with ammonium persulfate and N,N,N′,N′-tetramethylethylenediamine (TMEDA). The reaction could tolerate air, but was conducted under nitrogen to minimize radical termination reactions by oxygen. The reaction was completed within 30 min at room temperature. To ensure that all monomers and cross-linkers were incorporated into the polymer, the reaction mixture was allowed to stand under nitrogen at room temperature for another 30 min. The acrylated full-length sequence 4 was incorporated into the polymer to give the ODN–polymer conjugate 6 (Scheme 2). The failure sequences 5 and the impurities resulting from protecting groups remained in the polymer matrix, which were conveniently removed by extraction with water. The extracts were analyzed with RP HPLC to give trace b (Fig. 1). Compared with trace a, trace b did not have a peak at 62 min, which indicated that the full-length sequence was incorporated into polymer 6 efficiently.
 | ||
| Scheme 2 Purification of 5′-phosphorylated ODN through catching full-length sequence by polymerization. | ||
The ODN–polymer conjugate 6 was dried under vacuum. The intermediate ODN 7 was cleaved from polymer with HF–pyridine complex in DMF. Excess HF was quenched with Me3SiOMe. The supernatant, which contained the ODN 7, excess Me3SiOMe, Me3SiF, (iPr)2SiF2, MeOH, and the solvent DMF, was removed. The polymer was washed with water. The supernatant and the washes were combined and evaporated to dryness leaving only the ODN 7. To remove the tag on the 5′-phosphate group, to 7 was added concentrated ammonium hydroxide solution and heated to 80 °C for 30 min. Pure 5′-phosphorylated ODN 8 was obtained conveniently by direct nBuOH precipitation from the solution. As shown in HPLC trace c (Fig. 1), only one peak with a retention time of 19 min was observed. The recovery yield of the purification process was estimated to be 55% by comparing the area of the peak at 19 min in trace c with that of the peak at 62 min in trace a. The identity of the ODN was confirmed by MALDI-TOF MS analysis.
To further demonstrate the usefulness of this chemical phosphorylation and purification technology, the 61-mer ODN 9, which is a portion of HIV protease gene, was synthesized and phosphinylated with phosphoramidite 1. To increase the yield of the synthesis, controlled pore glass (CPG) with a pore size of 2000 Å was used. Before starting the automated synthesis, the CPG was manually capped with Pac2O for 20 min on the synthesizer. In addition, in the synthetic cycle, a 25-second waiting step was added after each delivery of coupling reagents (phosphoramidite and tetrazole) to the synthesis column. The capping step was performed two times. Following each capping step, a 50-second waiting step was added. Except for these modifications, the same synthesis procedure for ODN 4 was followed. The crude 9 was analyzed with RP HPLC (trace d, Fig. 1), and purified with the catching by polymerization procedure as described for purification of 4. The purified 61-mer ODN 10 was analyzed with RP HPLC to give trace e. The recovering yield was estimated to be 65% by comparing the area of the peak at 20 min in trace e with that of the peak at 54 min in trace d.

To avoid an extra heating step during deprotection of ODN, initially we used the UltraMild conditions for the synthesis of ODNs 4 and 9. However, in the more widely used base protecting strategy in DNA synthesis, dA is protected with a benzoyl group and dG is protected with an isobutyryl group. These protecting groups require heating in concentrated ammonium hydroxide for 8 h to remove. To test if our chemical phosphorylation and catching by polymerization techniques are compatible with these relatively harsher conditions, the 25-mer ODN 11 was synthesized using this normal protecting strategy. Specifically, phosphoramidite monomers were Bz-dA, iBu-dG, Ac-dC and dT. The capping reagent was acetic anhydride. Except for these modifications, all other reagents and conditions including the synthesis cycle are the same as those for the synthesis of 4. ODN 11 was cleaved from CPG with concentrated ammonium hydroxide at room temperature. The solution was then heated to 55 °C for 8 h to remove protecting groups. The crude 11 was analyzed with HPLC (trace f, Fig. 1) and purified with the catching by polymerization procedure as described for purification of 4. The purified 25-mer ODN 12 was analyzed with HPLC to give trace g. The recovering yield was estimated to be 79% by comparing the area of the peak at 18 min in trace g with that of the peak at 58 min in trace f.
This new ODN phosphorylation and purification technology provides a convenient method to access 5′-phosphorylated ODNs. We have found that the acrylation phosphoramidite 1 is stable for at least one month when stored at −20 °C under nitrogen in the dark. In the future, when this compound or its revised version is commercialized, highly pure 5′-phosphorylated ODNs can be obtained using the following procedure: (1) Coupling the acrylation phosphoramidite to the ODN at the end of synthesis. (2) Perform cleavage and deprotection as usual. (3) Subject the crude ODN to polymerization. (4) Wash the polymer with water. (5) Cleave ODN from the polymer. (6) Treat ODN with concentrated ammonium hydroxide shortly and precipitate ODN with nBuOH. Several advantages of this technology are remarkable. When compared with HPLC, this technology requires minimum volume of solvent; the cost of the HPLC instrument and its maintenance fee can also be saved. When compared with fluorous affinity purification and biotin–streptavidin enabled affinity purification methodologies, which are also efficient and convenient, this new technology does not require any solid phase extraction material. The chemicals for the polymerization reaction are all commercially available, inexpensive, and can be stored for extended periods of time under suitable conditions. These materials are only needed in small quantities. The most significant advantage of the new technology is that it can be readily scaled up. Because there is no need of any type of chromatography, and purification is achieved by simple manipulations such as shaking and extraction, large quantities of ODN can be purified in each batch. This can be easily envisioned by the fact that there is virtually no cost difference between purification of 1 mg of ODN and purification of 1 g of ODN. If any other methods are used, the difference could be enormous. Due to these advantages, we expect that the technology presented in this paper will be widely used in industry and academia for the production of pure 5′-phosphorylated ODNs.
Experimental
General
All reactions were performed in oven-dried glassware under a nitrogen atmosphere using standard Schlenk techniques. Reagents and solvents available from commercial sources were used as received unless otherwise noted. CH2Cl2 was distilled over CaH2. Thin layer chromatography (TLC) was performed using Sigma-Aldrich TLC plates, silica gel 60F-254 over glass support, 0.25 μm thickness. Flash column chromatography was performed using Selecto Scientific silica gel, particle size 32–63 μm. 1H, 13C and 31P NMR spectra were measured on a Varian UNITY INOVA spectrometer at 400, 100 and 162 MHz, respectively; chemical shifts (δ) were reported in reference to solvent peaks (residue CHCl3 at δ7.24 ppm for 1H and CDCl3 at δ77.00 ppm for 13C) and H3PO4 (at δ0.00 ppm for 31P). High-resolution mass spectra were obtained on a Finnigan Mat 95XL spectrometer. MALDI-TOF mass spectrum was obtained on a Shimadzu Biotech Axima CFRplus spectrometer. ODNs were synthesized on an ABI 394 solid phase synthesizer. HPLC was performed on a JASCO LC-2000Plus System, Pump PU-2089Plus Quaternary Gradient Pump, Detector UV-2075Plus. C-18 RP analytical column (5 μm diameter, 100 Å, 250 × 3.20 mm) was used. Solvent A: 0.1 M triethylammonium acetate, 5% acetonitrile; solvent B: 90% acetonitrile. All profiles were generated by detection of absorbance of DNA at 260 nm using the linear gradient solvent system: solvent B (0–45%) in solvent A over 60 min followed by solvent B (45%–100%) in solvent A over 20 min at a flow rate of 0.5 mL min−1. THF/pyridine/Pac2O, THF/pyridine/Ac2O, succinic ester linked DMTr-dT-lcaa-CPG (pore sizes 1000 Å and 2000 Å; lcaa = long chain alkyl amino), 5′-DMTr 2-cyanoethyl phosphoramidites acetyl-dC, Pac-dA (Pac = phenoxyacetyl), Bz-dA, 4-isopropyl-Pac-dG, iBu-dG and dT, and other commonly used solid phase DNA synthesis reagents were purchased from Glen Research, Inc.Synthesis of compound 3
A round-bottomed flask containing compound 216 (200 mg, 0.49 mmol, 1.0 equiv) and a magnetic stirring bar was evacuated and then refilled with nitrogen. The evacuation and nitrogen-filling cycle was repeated for two more times. Dry DMF (2 mL) and diisopropylethylamine (254 μL, 1.46 mmol, 3.0 equiv) were added via syringes. The mixture was cooled to 0 °C. Diisopropylsilyl bis(trifluoromethanesulfonate) (144 μL, 0.487 mmol, 1.0 equiv) in dry DMF (1 mL) was added via a syringe in one portion at 0 °C. The solution was stirred at 0 °C for 1 h and rt for 2 h. Imidazole (50 mg, 0.73 mmol, 1.5 equiv) in dry DMF (1 mL) was added via a syringe. The solution was stirred for 1 h, and then added to a flask containing diethyl bis(hydroxymethyl)malonate (107 mg, 0.487 mmol, 1.0 equiv), imidazole (33 mg, 0.487 mmol, 1 equiv) and DMF (2 mL) at 0 °C via a cannula slowly. The reaction mixture was stirred at 0 °C for 4 h, and then quenched with 5% NaHCO3 (0 °C, 50 mL). EtOAc (0 °C, 30 mL) was added, and the phases were separated. The aqueous phase was extracted with EtOAc (0 °C, 30 mL × 3). The combined organic phase was dried over anhydrous Na2SO4 and filtered. The filtrate was evaporated under reduced pressure to give a yellow oil. Purification with flash column chromatography (SiO2, hexanes/EtOAc, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) gave 3 as a pale yellow oil (151 mg, 42%): Rf 0.60 (SiO2, hexanes/EtOAc, 1:2); 1H NMR (400 MHz, CDCl3) δ 6.07 (br s, 1H), 5.94 (br s, 1H), 5.614–5.609 (m, 1H), 5.25–5.24 (m, 1H), 4.23 (s, 2H), 4.21–4.08 (m, 6H), 3.86 (br s, 1H), 3.26–3.21 (m, 2H), 3.18–3.13 (m, 2H), 2.26–2.14 (m, 2H), 1.91–1.90 (m, 3H), 1.83–1.77 (m, 2H), 1.53–1.37 (m, 8H), 1.25–1.19 (m, 22H), 0.99–0.95 (m, 14H), 0.83–0.76 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 173.8, 169.3, 168.7, 140.4, 119.3, 78.8, 61.8, 61.6, 61.5, 61.4, 39.9, 39.8, 34.7, 31.6, 31.3, 31.1, 30.9, 29.7, 29.6, 29.4, 27.1, 18.9, 18.1, 17.9, 14.4, 14.2, 13.7, 8.5; HRMS (ESI, [M+Na]+) calcd for C39H74N2NaO9Si 765.5061, found 765.5069.
:1 to 1:1) gave 3 as a pale yellow oil (151 mg, 42%): Rf 0.60 (SiO2, hexanes/EtOAc, 1:2); 1H NMR (400 MHz, CDCl3) δ 6.07 (br s, 1H), 5.94 (br s, 1H), 5.614–5.609 (m, 1H), 5.25–5.24 (m, 1H), 4.23 (s, 2H), 4.21–4.08 (m, 6H), 3.86 (br s, 1H), 3.26–3.21 (m, 2H), 3.18–3.13 (m, 2H), 2.26–2.14 (m, 2H), 1.91–1.90 (m, 3H), 1.83–1.77 (m, 2H), 1.53–1.37 (m, 8H), 1.25–1.19 (m, 22H), 0.99–0.95 (m, 14H), 0.83–0.76 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 173.8, 169.3, 168.7, 140.4, 119.3, 78.8, 61.8, 61.6, 61.5, 61.4, 39.9, 39.8, 34.7, 31.6, 31.3, 31.1, 30.9, 29.7, 29.6, 29.4, 27.1, 18.9, 18.1, 17.9, 14.4, 14.2, 13.7, 8.5; HRMS (ESI, [M+Na]+) calcd for C39H74N2NaO9Si 765.5061, found 765.5069.
Synthesis of phosphoramidite 1
A round-bottomed flask containing 3 (119 mg, 0.16 mmol, 1.0 equiv) and a magnetic stirring bar was evacuated and then refilled with nitrogen. The evacuation and nitrogen-filling cycle was repeated for two more times. Dry CH2Cl2 (5 mL) and 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphoramidite (60 μL, 0.176 mmol, 1.1 equiv) were then added via syringes. A 1H-tetrazole solution in CH3CN (0.45 M, 391 μL, 0.176 mmol, 1.1 equiv) was added via a syringe in one portion. After stirring at rt for 2 h, the reaction mixture was concentrated to dryness under reduced pressure. The residue was purified with flash column chromatography (SiO2, hexanes/EtOAc/Et3N = 3:1:1) giving 1 as a colorless oil (150 mg, 99%): Rf 0.40 (SiO2, hexanes/EtOAc/Et3N = 3:1:1); 1H NMR (400 MHz, CDCl3) δ 6.00 (br s, 1H), 5.85 (br s, 1H), 5.64–5.60 (m, 1H), 5.263–5.257 (m, 1H), 4.23–4.10 (m, 8H), 3.80–3.71 (m, 2H), 3.56–3.47 (m, 2H), 3.28–3.23 (m, 2H), 3.20–3.15 (m, 2H), 2.58–2.55 (m, 2H), 2.27–2.15 (m, 2H), 1.921–1.920 (m, 3H), 1.85–1.74 (m, 2H), 1.56–1.38 (m, 8H), 1.24–1.18 (m, 24H), 1.14–1.11 (m, 10H), 1.00–0.98 (m, 14H), 0.82 (t, 6H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 173.6, 168.6, 140.5, 119.2, 117.7, 79.1, 61.7, 61.5, 61.1, 61.0, 58.7, 58.5, 43.4, 43.3, 39.9, 39.7, 35.6, 34.2, 31.9, 31.1, 29.8, 29.71, 29.67, 29.59, 29.50, 29.46, 27.1, 24.8, 24.74, 24.69, 24.66, 20.55, 20.49, 18.9, 18.1, 17.9, 14.4, 14.1, 13.6, 8.53; 31P NMR (162 MHz, CDCl3) δ 149.9.
ODN 4 synthesis, cleavage and deprotection
The 20-mer 5′-acrylated and phosphorylated ODN 4 was synthesized on an ABI DNA/RNA synthesizer at 1 μmol scale. The solid support was lcaa-CPG with a pore size of 1000 Å. The ODN was anchored to the support through a succinic ester linkage. The following 5′-DMTr-protected 2-cyanoethyl phosphoramidite monomers were used for the synthesis: Pac-dA, 4-isopropyl-Pac-dG, Ac-dC and dT. THF/pyridine/Pac2O was used as the capping reagent. The manufacturer recommended synthetic cycle was followed except that the phosphoramidite 1 was coupled for 5 min. After synthesis, the ODN was cleaved from CPG with concentrated NH4OH at rt and allowed to stand under these conditions for 8 h. The solution of crude ODN was divided into 10 equal portions, and evaporated to dryness in 10 Eppendorf tubes in a vacuum SpeedVac concentrator. One portion was dissolved in 150 μL water, of which 20 μL was injected into HPLC to generate trace a (Fig. 1).Polymerization of full-length ODN 4
The remaining 130 μL solution of the crude ODN 4 was transferred into a 2-necked round-bottomed flask. The Eppendorf tube was washed with water (40 μL × 3) and the washes were added to the flask. A polymerization solution [250 μL; dimethylacrylamide (1.69 M) and N,N′-methylenebis(acrylamide) (16.9 mM) in water; the solution can be pre-prepared and stored at −20 °C in the dark for at least 1 month] was added via a pipette. The mixture was gently stirred under a nitrogen flow for 2 min. The solution of (NH4)2S2O4 (10%, 5 μL) was then added via a pipette, which was followed by N,N,N′,N′-tetramethylethylenediamine (TMEDA, 5 μL). The mixture was stirred gently under nitrogen at rt for 30 min. The ODN–polyacrylamide conjugate 6 was formed. The failure sequences 5 remained in solution (Scheme 2). The gel was allowed to stand for another 30 min to ensure complete polymerization.Removal of failure sequences and other impurities
To the ODN–polymer conjugate 6 in the round-bottomed flask was added 3 mL water. The content was gently shaken at rt overnight. The supernatant, which contained the failure sequences 5 and other impurities, was removed with a pipette. The gel was further washed with water (2 mL × 3; 2 h each time). The supernatant and the washes were combined and evaporated to dryness. The residue was dissolved in 130 μL water, of which 20 μL was injected into HPLC to generate trace b (Fig. 1).Cleavage of full-length ODN from polymer and releasing the 5′-phosphate group
The gel in the round-bottomed flask was dried under vacuum. Dry DMF (2 mL) was added via a pipette, which was followed by HF–pyridine complex (60 μL). The mixture was shaken gently under nitrogen for 5 h. Me3SiOMe (500 μL) was then added. After shaking for 15 min, the supernatant was transferred to Eppendorf tubes. The gel was extracted with water (2 mL × 3 at rt; 12 h, 2 h, 2 h, respectively). The supernatant and the extracts were evaporated to dryness in a SpeedVac vacuum concentrator and were combined to give ODN 7 (Scheme 2). To deprotect the 5′-phosphate group of 7, concentrated NH4OH (∼28%, 100 μL) was added. After a short vortex, the mixture was heated to 80 °C for 30 min. After cooling to rt, nBuOH (900 μL) was added. The mixture was vortexed for 30 s and then centrifuged at 14.5 K for 5 min. The supernatant was removed. The residue was further dried shortly in a SpeedVac. The ODN 8 was dissolved in 130 μL water, of which 20 μL was injected into HPLC to generate profile c (Fig. 1). The recovery yield of the purification process was estimated to be 55% by comparing the area of the peak in trace c at 19 min with that in trace a at 62 min. MALDI-TOF mass spectrum of ODN 8: calcd for [M−2H+Na]− C194H247N67NaO125P20 6159.0, found 6159.8.Synthesis and purification of the 61-mer ODN 10
For the solid phase synthesis, cleavage and deprotection of 9, the procedure for 4 was followed except for the following modifications. CPG with a pore size of 2000 Å instead of 1000 Å was used. Before synthesis, the CPG was manually capped with Pac2O for 20 min on the synthesizer. In the synthetic cycle, a 25-second waiting step was added after each delivery of coupling reagents (phosphoramidite and tetrazole) to the synthesis column. An additional capping step was added, and after each capping step, a 50-second waiting step was added. The catching by polymerization procedure was exactly the same as described for 4. However, for RP HPLC analysis of 10, buffer A that contained 10% urea was used.Synthesis and purification of the 25-mer ODN 12 using normal base protecting groups
For the solid phase synthesis, cleavage and deprotection of 11, the procedure for 4 was followed except for the following modifications. The 5′-DMTr-protected 2-cyanoethyl phosphoramidite monomers Bz-dA, iBu-dG, Ac-dC and dT were used. The capping agents were replaced with THF/pyridine/Ac2O. After cleaving the ODN from CPG with concentrated NH4OH at rt, the solution was heated to 55 °C for 8 h in a tightly capped vial. The catching by polymerization procedure was exactly the same as described for 4.Conclusions
In conclusion, by combining a commercialized chemical phosphorylation technique and our newly developed ODN purification methodology, we have developed a new technology for the purification of synthetic 5′-phosphorylated ODN. The technology is simple, convenient, inexpensive, and highly efficient. It may be readily scaled up and affords pure ODN at both small and large scales. We believe that it will provide a more affordable way for scientists to obtain 5′-phosphorylated ODNs for applications in chemistry, biology and medicine.Acknowledgements
Financial support from US NSF (CHE-0647129), Michigan Universities Commercialization Initiative, MTU Research Excellence Fund (REF-TC), MTU Chemistry Department, MTU Biotech Research Center, and The Royal Thai Government Scholarship (S. F.); the assistance from Mr Jerry L. Lutz (NMR), Mr Shane Crist (computation), and Mr Dean W. Seppala (electronics); and an NSF equipment grant (CHE-9512445) are all gratefully acknowledged.References
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151–164 CrossRef CAS.
- G. De Rosa and M. I. La Rotonda, Molecules, 2009, 14, 2801–2823 CrossRef CAS.
- C.-Z. Zhou and J. Chattopadhyaya, Curr. Opin. Drug Discovery Dev., 2009, 12, 876–898 CAS.
- M. Khati, J. Clin. Pathol., 2010, 63, 480–487 CrossRef CAS.
- Y. Singh, P. Murat and E. Defrancq, Chem. Soc. Rev., 2010, 39, 2054–2070 RSC.
- D.-Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS.
- A. Matsuda, J. Pharmaceut. Soc. Japan, 2011, 131, 285–298 CAS.
- X. Yang, N. Li and D. G. Gorenstein, Expert Opin. Drug Discovery, 2011, 6, 75–87 CrossRef CAS.
- Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley & Sons, New York Search PubMed.
- W. H. Pearson, D. A. Berry, P. Stoy, K. Y. Jung and A. D. Sercel, J. Org. Chem., 2005, 70, 7114–7122 CrossRef CAS.
- C. Beller and W. Bannwarth, Helv. Chim. Acta, 2005, 88, 171–179 CrossRef CAS.
- S. Fang and D. E. Bergstrom, Tetrahedron Lett., 2004, 45, 7987–7990 CrossRef CAS.
- S. Fang and D. E. Bergstrom, Nucleic Acids Res., 2003, 31, 708–715 CrossRef CAS.
- S. Fang and D. E. Bergstrom, Bioconjugate Chem., 2003, 14, 80–85 CrossRef CAS.
- B. S. Sproat, T. Rupp, N. Menhardt, D. Keane and B. Beijer, Nucleic Acids Res., 1999, 27, 1950–1955 CAS.
- S. Fang and S. Fueangfung, Org. Lett., 2010, 12, 3720–3723 CrossRef CAS.
- S. Fang, S. Fueangfung, X. Lin, X. Zhang, W. Mai, L. Bi and S. A. Green, Chem. Commun., 2011, 47, 1345–1347 RSC.
- P. Modrich and I. R. Lehman, J. Biol. Chem., 1973, 248, 7502–7511 CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning: A Laboratory Manual, 2001, Cold Spring Harbor Press, Cold Spring Harbor, New York Search PubMed.
- H.-J. Fritz, in DNA Cloning: A Practical Approach, ed. D. M. Glover, IRL Press, Oxford, 1985, vol. 1, pp. 151–163 Search PubMed.
- F. Barany, PCR Methods and Applications, 1991, 1, 5–16 CAS.
- T. Horn and M. S. Urdea, Tetrahedron Lett., 1986, 27, 4705–4708 CrossRef CAS.
- A. Guzaev, H. Salo, A. Azhayev and H. Lonnberg, Tetrahedron, 1995, 51, 9375–9384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and 31P NMR spectra of new compounds; MALDI-TOF mass spectrum of ODN 8. See DOI: 10.1039/c2ra01357f |
| This journal is © The Royal Society of Chemistry 2012 |