Convertible electron transfer pathways of cytochrome c at TiO2 quantum electrode
Li
Liu
,
Ning
Wang
* and
Lin
Guo
*
Key Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry and Environment, Beijing University of Aeronautics and Astronautics, Beijing, 100191, People's Republic of China. Tel: 86-10-82338162E-mail: wangning@ buaa.edu.cn; guolin@buaa.edu.cn.
First published on 8th February 2012
Abstract
In this work, biofunctional ultrathin TiO2 quantum wires (about 3 nm), self-assembled into flat-cable-like “stacks”, are synthesized by a simple template-free wet chemical method with high uniformity and on a large scale. On the self-assembled TiO2 nanowire (SA-Nw-TiO2) coated electrode, cytochrome c (cyt c) undergoes a fast, reversible (or quasi-reversible) and sensitive electron transfer process that is closely related to the relative ionic strength and the hydrated state of SA-Nw-TiO2 surface. At the suitably hydrated SA-Nw-TiO2 electrode in an optimum bulk ionic environment, electrons are reversibly transferred at the heme edge on the basis of an effective orientation of cyt c. It is interesting to note that in a suitable ionic microenvironment, a convertible electron transfer occurs in the following three steps: electron transfer driven partial unfolding of cyt c, electron transfer, and thermodynamics driven refolding. In addition, the partially unfolded cyt c was proven to exist only in the quasi-reversible electron transfer process, and the multi-step pathway of electron transfer can be converted into the single-step pathway at the fully hydrated SA-Nw-TiO2 electrode.
Introduction
Cytochrome c (cyt c) is a basic redox heme protein, which plays an important role in the biological respiratory chain by transferring electrons between two inner-membrane-bound proteins, cyt c reductase and cyt c oxidase.1,2 Generally, cyt c is a small soluble globular protein (2.6 nm × 3.0 nm × 3.2 nm) with the heme covalently bounded by two thioether bridges to Cys14 and Cys17 and axially liganded with His18 and Met80.3 To form these bonds, the heme is embedded in the hydrophobic interior of the globular polypeptide, leaving an edge exposed to solvent. The polypeptide must enclose the heme moiety on at least three sides and form a crevice structure.4 Up to date, it has been disclosed that the electron transport process is closely related to the intrinsic conformational flexibility of cyt c mentioned above, which is obviously affected by environmental conditions such as pH, temperature and ionic strength.5,6 Among them, ionic strength has a greater significant effect on the rate and potential of the electron transfer through changing the cyt c surface charge or inducing the reorientation of cyt c,7–9 which then affects the physiological functions of the protein and the applications in catalysis or as biosensors.In recent years,10,11 TiO2-based semiconductor nanostructures have been attracting a great amount of interest due to their potential use as electrode materials for biointerfacial electron transfer or biosensing on the basis of the bioactivity, signal amplification effect and environmental safety.12–15 TiO2 quantum wires and their assemblies are ideal platforms for the investigation of the direct electrochemistry of cyt c, owing to their significant enhancement on adsorption and biocompatibility for the targeted protein based on their large specific surface area, highly organized nanostructures and mesopores, which resulted in higher bio-sensitivity and faster sensing response.16,17 However, in contrast with the measurements made along with the increase of ionic strength in big strides reported before,7–9 research on the effect of minor fluctuations in ionic microenvironment on the electron transfer process of cyt c at a nanoscale TiO2 electrode is rather limited, although such a work may present a deeper understanding of some regional phenomena such as critical conformational fluctuations as well as electron pathways conversion.17 New insight into the interplay of suitable strength of electrostatic attraction between cyt c and electrode (EA) and the corresponding hydrophobic interaction (HI) is only of scientific interest in the electron transfer kinetics of cyt c but also could provide real benefits such as the development of related biosensors and biomaterials.
Thus in this work, a simple template-free wet chemical method was first utilized to synthesize flat-cable-like “stacks” of ultrathin (about 3 nm) rutile TiO2 nanowires with high uniformity and on a large scale. Research into the relative ionic strength effect on the direct electrochemistry of cyt c was then carried out on the self-assembled TiO2 nanowire (SA-Nw-TiO2) coated electrode. It is found that the electrochemical behavior of cyt c is closely related to the relative ionic strength and the hydrated state of SA-Nw-TiO2 surface. At a suitably hydrated SA-Nw-TiO2 electrode interface, cyt c in a suitable ionic microenvironment could show a redox response of partially unfolded form, and form a new stable electron transfer pathway. Moreover, it was also found that the new pathway of electron transfer can be re-converted into the conventional pathway by modulating the hydrated state of the SA-Nw-TiO2 surface, which was also closely related to the unique hierarchical structure of the flat-cable-like TiO2 stacks.
Experimental
Sample synthesis
In a typical synthesis process, 10 ml pure TiCl4 was first dissolved in 40 ml pure water and then mixed with 12.5 ml 2 M NaOH solution under vigorous agitation. The mixture was then refluxed for 1 h. White precipitation occurred immediately as soon as the solution was refluxed. After being washed with pure water and ethanol three times respectively, the titania powders were dried at 60 °C.Characterization
Characterization of the produced materials was made by X-ray diffraction (XRD) (Rigaku Goniometer PMG-A2, CN2155D2), transmission electron microscopy (TEM) (JEOL 200 CX) and field emission scanning microscopy (Hitachi S4800). UV-vis spectra were recorded with a UV-vis spectrophotometer (GBC Cintra 10e).Electrochemical measurements were performed with a CHI 660C Electrochemical Analyzer (CH instrument, USA) in a conventional three-electrode cell. Coated glassy carbon (GC) electrodes were used as the working electrodes, a platinum wire as the counter electrode and an aqueous Ag/AgCl as the reference electrode. A GC electrode (3 mm diameter) was polished with 1.0, 0.3, and 0.05 μm alumina slurry sequentially, and then washed and ultrasonicated in water and then ethanol for a few minutes. To accomplish the preparation of the coated electrodes, 1 mg as-prepared TiO2 nanowires was dispersed into 1 ml ethanol and then sonicated for 1 h until a homogeneous suspension resulted. 10 μl of the 1 mg/ml TiO2 nanowires suspension was spread evenly onto the treated GC electrode surface with a syringe. In order to obtain uniform films, the coated electrodes were covered with a small beaker and allowed to dry for over 24 h at room temperature. The coated electrodes were immersed in electrolyte for a day to fabricate suitably hydrated electrodes. The electrolyte was 25 mM phosphate buffer solution (PBS) at pH 6.8 made from analytical grade Na2HPO4 and KH2PO4. Horse heart muscle cyt c was purchased from Acros and was used without purification.
Results and discussion
1. Structural characterization


2. Convertible electron transfer pathways of cyt c on the self-assembled TiO2 nanowire (SA-Nw-TiO2) coated electrode
 | ||
| Fig. 3 CVs of 0.72 and 0.02 mg/ml cyt c PBS (25 mM, pH 6.8) at the suitably hydrated SA-Nw-TiO2 coated electrode at 5 mV s−1. | ||
For the native cyt c, the cathodic peak current was proportional linearly to the square root of the scan rates (Fig. 4), suggesting a diffusion-controlled electrode process.20 In the range 1–20 mV s−1, ΔE changed between 59 and 74 mV. At 5 mV s−1, the heterogeneous electron transfer rate constant (k0) of native cyt c can be calculated as 1.3 × 10−2 cm s−1 according to the Nicholson equation, confirming that the native response corresponds to a reversible electrode process.21
 | ||
| Fig. 4 CVs of 0.72 mg/ml cyt c PBS at the suitably hydrated SA-Nw-TiO2 electrode with various scan rates: 1, 2, 5, 10 and 20 mV s−1. Inset: Plots of cathodic peak currents vs. the square root of scan rates. | ||
In previous work, it has been proposed that a combination of electrostatic and chemical interaction at the electrode–solution interface determines the heterogeneous electron transfer of cyt c.22 Cyt c is a highly ionic protein that carries a net charge of +7/+8 at pH 7.23 Many anions, such as phosphate ions, bind to the positively charged lysine residues surrounding the exposed heme edge and cause a reduction in the net positive charge of cyt c.24 The specific ion binding to the lysine residues in the active site region also results in an opening of the heme crevice and an solution exposure of the heme edge (Ohc&SEhe) without alteration of heme's coordination configuration.25 For the rutile SA-Nw-TiO2 (pI 5.7), a solution of pH 6.8 should produce a negatively charged surface. Clean SA-Nw-TiO2 surfaces are generally thought to have an inner monolayer of ordered water molecules and a second monolayer of hydrated electrolyte ions which should be packed in less order.22
At 0.72 mg/ml cyt c, the fast and reversible native response comes from suitable strength of rapid adsorption of cyt c, effective orientation, and rapid desorption on or from the SA-Nw-TiO2 coated electrode. The electrostatic attraction occurred between positively charged lysine residues surrounding exposed heme edge and negatively charged oxygen atoms resulted from dissociation of SA-Nw-TiO2 surface hydroxyls or the hydrated phosphate ions initially adsorbed on the SA-Nw-TiO2 surface (EA). Nevertheless, the EA would decrease when partly shielded by the formation of compact ionic atmospheres around cyt c. At such a short interaction distance, there also occurred the hydrophobic interaction between hydrophobic groups near the heme edge which are exposed upon Ohc&SEhe and some hydrophobic sites on the SA-Nw-TiO2 surface (HI), as well as the hydrogen binding of the lysine residues to oxygen atoms of hydration layer water (HB). During the potential scanning, EA, HI and HB can effectively compensate for the electrostatic repulsion from the positive electric field (ER). As a result, the suitable strength of rapid adsorption can hold cyt c into the proper orientation on the SA-Nw-TiO2 electrode for effective electron transfer, which is believed to occur at the heme edge of native cyt c.3 On reduction, the heme moves into the hydrophobic interior of the protein sheath, and the heme crevice closes. The movements may help the rapid desorption of cyt c from the electrode surface and the following electron transfer.25 On re-oxidization, a stronger ER during the anodic scanning leads to the rapid desorption of cyt c, as confirmed by the diffusion-controlled electrode process of native cyt c.
2.2 Electron transfer of partially unfolded cyt c
UV-vis absorption spectra in Fig. 5 show that 0.01, 0.02 and 0.05 mg/ml cyt c in 25 mM PBS (pH 6.8) exhibited a Soret band at 408 nm and Q band at 528 nm, suggesting a native conformation of cyt c in the three solutions.26 However, different electrochemical behaviors were observed, indicating that the partially unfolded cyt c was formed at the SA-Nw-TiO2 electrode–solution interface, closely related to the difference in the heme's ionic microenvironment being delicately affected by the change of ionic strength relative to cyt c concentration (I/C). Notably, at 0.03 mg/ml, Soret and Q bands showed a hypsochromic shift to 404 and 524 nm with the full width at half-maximum (HWHM) broadening, and another weak Q-band appeared at ca. 622 nm. At 0.04 mg/ml, a further hypsochromic shift to 395 and 521 nm occurred with the HWHM and the new Q band further broadening and increasing in intensity. According to the exciton theory, a hypsochromic shift and band broadening of Soret band indicate a characteristic of excitonic coupling between porphyrins as the distance between the chromophores decreases.27 Thus, cyt c underwent partial unfolding and then self-aggregation in 0.03 mg/ml cyt c PBS, and larger aggregates were formed in 0.04 mg/ml cyt c PBS. The appearance of an additional Q-band may be associated with a vibrationally accessible mode of the aggregates.28 As the lyophilized sample was added into 0.04 mg/ml cyt c PBS, a disaggregation and then refolding into native monomers occurred in 0.05 mg/ml cyt c PBS. Concentration-modulated self-aggregation and disaggregation indicate that the partial unfolding and refolding of cyt c can be reversible in the electrolyte under the combination of a proper ionic microenvironment and adequate cyt c concentration.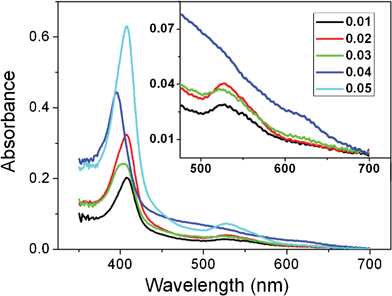
For the partially unfolded cyt c, both cathodic and anodic peak currents were found to be proportional linearly to the square root of the scan rates (Fig. 6), suggesting a diffusion-controlled electrode process.20 In the range 1–20 mV s−1, ΔE increased from 69 to 88 mV. At 5 mV s−1, k0 of partially unfolded cyt c can be calculated to be 1.1 × 10−3 cm s−1. The data confirm that a partially unfolded response corresponds to a quasi-reversible process.
 | ||
| Fig. 6 CVs of 0.02 mg/ml cyt c PBS (25 mM, pH 6.8) at the suitably hydrated SA-Nw-TiO2 electrode with various scan rates: 1, 2, 5, 10 and 20 mV s−1. Inset: Plots of cathodic (upper) and anodic (lower) peak currents vs. the square root of scan rates. | ||
The partially unfolded response is a result of the interplay of EA, HI, HB and ER between cyt c in the suitable ionic microenvironment and the suitably hydrated SA-Nw-TiO2 electrode. As I/C rise, the net positive charge of cyt c decreases while the degree of Ohc&SEhe increases; correspondingly, the ionic microenvironment around cyt c changes. In such an ionic microenvironment, EA decreases with the net positive charge decreasing and thus the interaction distance between cyt c and SA-Nw-TiO2 increases; HI and HB decrease with the increase in interaction distance but HI also increases with the increase in the degree of Ohc&SEhe. In the case of 0.02 mg/ml cyt c PBS, the suitable ionic microenvironment could balance the two opposing contributions of HI to take it to the maximum. To transfer electrons at a more positive potential, the strong enough HI could expand itself through further extending the Ohc&SEhe so that the further enhanced HI, together with EA and HB, could compensate for the strong ER at this potential. As a result, the Met80-S-iron linkage was disrupted and a partially unfolded cyt c responded on the suitably hydrated SA-Nw-TiO2 electrode. Here, the heme lay flat on the electrode and the rapid electron transfer could occur directly between the iron atom and electrode, but not through the porphyrin ring. The directness of the iron atom's electron transfer, together with the absence of steric hindrance and the shortening of response distance made the electrochemical behavior of partially unfolded cyt c similar to that of Fe(CN)63−/Fe(CN)64−, thus Ef positively shifts to 0.22 V. Due to thermodynamic factors that the reduced state of native cyt c is particularly stable and that the re-oxidized state of cyt c suffers from a stronger and stronger ER, the partially unfolded cyt c rapidly disengaged from the electrode surface with the Met80-S-iron relinked following electron transfer. Thus, the whole electrode process appears as diffusion-controlled. The procedures of partial unfolding and subsequent refolding enable the electrochemistry of partially unfolded cyt c to be quasi-reversible instead of reversible. The current intensity of 0.02 mg/ml cyt c at 0.22 V is equivalent to that of 0.72 mg/ml cyt c at 0.074 V, which is in accordance with the proposal that the exchange current density of heme in cyt c would be 0.6% of that value for a free heme having identical reactivity.29 Also, there is a weak couple at ca. 0.074 V, which corresponds to a small amount of cyt c with not enough degree of Ohc&SEhe since the change of cyt c′ surface charges upon specific ion binding, is in the statistical sense.
2.3 Native and partially unfolded responses of cyt c in the whole concentration range
In the whole concentration range (Fig. 7), the transferred charges of native cyt c (Qn) in the cases of 0.01 to 0.04 mg/ml formed a plateau region with a very low charge value due to the substantial reduction of native cyt c molecules on the electrode or in the electrolyte. With the exception of the four concentrations, Qn increased at a decreasing rate with concentration from 0.0032 to 0.17 mg/ml. From 0.17 mg/ml on, the cathodic peak current of native cyt c (In) increased linearly with concentration and eventually deviated from the linearity at ca. 4 mg/ml. Thus it can be concluded that the TiO2 quantum electrode exhibited very high sensitivity for detection of cyt c. | ||
| Fig. 7 Concentration-dependent Qn or Qpu transferred at the suitably hydrated SA-Nw-TiO2 electrode: from 0.0032 to 0.17 mg/ml cyt c PBS (25 mM, pH 6.8). Inset: concentration-dependent In: from 0.17 to 6.23 mg/ml. | ||
At 0.01 mg/ml cyt c PBS (25 mM, pH 6.8), HI was lower than the maximum due to a longer interaction distance although with a greater degree of Ohc&SEhe. In this case, only a small amount of cyt c in this solution could exhibit a partially unfolded response in contrast with that at lower concentrations. From 0.0032 to 0.17 mg/ml cyt c PBS with the exception of 0.01 to 0.04 mg/ml, during the interplay of EA, HI, HB and ER, the degree of Ohc&SEhe may play an important role in producing the native redox response. In these ionic microenvironments, the greater the degree of Ohc&SEhe, the higher percent of the total amount of cyt c could be guided into an effective orientation on the SA-Nw-TiO2 electrode, causing a higher reactivity of cyt c. Above 0.17 mg/ml, cyt c could form a buffer system with 25 mM PBS (pH 6.8). With a net positive charge on cyt c, the degree of Ohc&SEhe and the ionic microenvironment show a slight change with cyt c concentration. The bulk ionic environment may play the key role in producing the native redox response and lead to a constant reactivity of cyt c. Thus, Qn (In) increased at a decreasing rate and then linearly with the increase of cyt c concentration.
2.4 Conversion of partially unfolded response into native response
Furthermore, it is also found that the redox response of partially unfolded cyt c can be converted into that of native cyt c at a fully buffer-soaked SA-Nw-TiO2 electrode as substituted for a freshly prepared one in response to 0.02 mg/ml cyt c PBS (Fig. 8). The fully buffer-soaked electrode was obtained by soaking a freshly prepared SA-Nw-TiO2 electrode in 25 mM PBS (pH 6.8) for a week. During the extended soaking in buffer, hydrophobic SA-Nw-TiO2 was fully wetted by water and the electrode–solution interfacial region attained equilibrium of hydration. Thus, it can be concluded that a stable well-behaved partially unfolded response stems from SA-Nw-TiO2 surface with suitable degree of hydration and cyt c in suitable ionic microenvironment. | ||
| Fig. 8 CVs of 0.02 mg/ml cyt c PBS at a freshly prepared SA-Nw-TiO2 electrode and at the SA-Nw-TiO2 electrode being soaked in 25 mM PBS (pH 6.8) for a week. | ||
At a freshly prepared SA-Nw-TiO2 electrode, rapid hydration in the earliest stage could make the partially unfolded response very unstable and too strong HI could induce the irreversible adsorption of cyt c on the electrode. At the fully buffer-soaked SA-Nw-TiO2 electrode, the fully hydrated electrode surface can modulate HB to a great extent so that the enhanced HB, together with EA and limited HI, could be strong enough to support cyt c (which should have been partially unfolded at a suitably hydrated SA-Nw-TiO2 electrode) into an effective orientation to transfer electron at the heme edge. At the suitably hydrated SA-Nw-TiO2 electrode, a new stable pathway of electron transfer for cyt c in suitable ionic microenvironment occurs: electron transfer driven partial unfolding of cyt c, electron transfer, and thermodynamics driven refolding. Moreover, the multi-step electron transfer pathway can be converted into the single-step pathway when the SA-Nw-TiO2 surface has been fully hydrated.
In summary, highly sensitive native response, reversible partial unfolding of cyt c, and conversion of partially unfolded to native response, are all ascribed to the sensitive SA-Nw-TiO2/25 mM PBS (pH 6.8) interface, which can rapidly and accurately respond to a minor change in the conformation of cyt c upon minor changes in the ionic microenvironment or the hydration state of SA-Nw-TiO2. The suitable interfacial microenvironment originates from not only the optimum bulk ionic strength30 but also the unique structure of SA-Nw-TiO2. Firstly, the parallel stacked microstructures result in the formation of two 3-D networks, one consisting of the flat-cable-like TiO2 nanowires and the other consisting of the interconnected nanopores, while the latter is also blended with the macropores of the flat cable coating. Such a stable nanostructure can greatly enhance the adsorption surface areas of cyt c and effectively promote the transportation and accessibility of cyt c to the SA-Nw-TiO2 coated electrode surface, which contributes to a high sensitivity. Secondly, the ultrathin TiO2 nanowires match well with cyt c in size. The quantum size, together with the sound crystallinity and high surface curvature, endows SA-Nw-TiO2 with a favorable distribution of surface charge, resulting in the appropriate electrostatic interaction with cyt c. Thirdly, the suitable strength of hydrophilicity and hydrophobicity can cause the favorable chemical interaction between SA-Nw-TiO2 and cyt c in the same order with the electrostatic interaction. As has been reported,31,32 the adjustability can serve the SA-Nw-TiO2/25 mM PBS (pH 6.8) interface as a bio-smart interface.
The bio-smart interface can modulate the contributions of EA and HI in response to a series of concentrations of cyt c in 25 mM PBS (pH 6.8), resulting in an Ohc&SEhe-controlled region and a bulk ionic environment-controlled region. The bio-smart interface can balance the two opposing contributions of HI to take it to the maximum, realizing the reversible partial unfolding of cyt c and a new stable electron transfer pathway. It can also enhance the contribution of HB in response to the extended soaking so as to induce a conversion of partially unfolded response into native response.
Conclusions
A novel electrode material, self-assembled TiO2 ultrathin nanowires (SA-Nw-TiO2), was synthesized by a simple template-free wet chemical method with high uniformity. In 25 mM PBS (pH 6.8), the SA-Nw-TiO2 coated electrode surface can serve as a sensitive bio-smart interface to rapidly and accurately respond to the minor changes in ionic microenvironment or the hydrated state of SA-Nw-TiO2. During the modulating process, the bio-smart interface accomplishes highly sensitive native response, reversible partial unfolding of cyt c and conversion of partially unfolded to native response. This sensitivity is assigned to the ability to subtly modulate the strength of electrostatic and chemical interaction with cyt c, eventually to the sound crystallinity, high surface curvature, suitable strength of hydrophilicity and hydrophobicity, quantum size and structural stability of SA-Nw-TiO2.Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2010CB934700) and the National Natural Science Foundation of China (Nos. 20973019, 21173017, 50902007 and 50725208).References
- J. Gong, P. Yao, H. Duan, M. Jiang, S. Gu and L. Chunyu, Biomacromolecules, 2003, 4, 1293 CrossRef CAS.
- J. Li, G. Cheng and S. Dong, J. Electroanal. Chem., 1996, 416, 97 CrossRef CAS.
- G. Moore, Z. Huang, C. Eley, H. Barker, G. Williams, M. Robinson and R. Williams, Faraday Discuss. Chem. Soc., 1982, 74, 311 RSC.
- P. George and R. Lyster, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 1013 CrossRef CAS.
- M. Fedurco, J. Augustynski, C. Indiani, G. Smulevich, M. Antalík, M. Bánó, E. Sedlák, M. Glascock and J. Dawson, J. Am. Chem. Soc., 2005, 127, 7638 CrossRef CAS.
- F. Sinibaldi, L. Fiorucci, A. Patriarca, R. Lauceri, T. Ferri, M. Coletta and R. Santucci, Biochemistry, 2008, 47, 6928 CrossRef CAS.
- A. Avila, B. Gregory, K. Niki and T. Cotton, J. Phys. Chem. B, 2000, 104, 2759 CrossRef CAS.
- Y. Sallez, P. Bianco and E. Lojou, J. Electroanal. Chem., 2000, 493, 37 CrossRef CAS.
- J. Petrović and R. Clark, Langmuir, 2005, 21, 6308 CrossRef.
- Y. Liu, J. Li, M. Wang, Z. Li, H. Liu, P. He, X. Yang and J. Li, Cryst. Growth Des., 2005, 5, 1643 CAS.
- J. Li and J. Zhang, Coord. Chem. Rev., 2009, 253, 3015 CrossRef CAS.
- Y. Zhang, P. He and N. Hu, Electrochim. Acta, 2004, 49, 1981 CrossRef CAS.
- E. Topoglidis, T. Lutz, J. Durrant and E. Palomares, Bioelectrochemistry, 2008, 74, 142 CrossRef CAS.
- G. Zhao, Y. Lei, Y. Zhang, H. Li and M. Liu, J. Phys. Chem. C, 2008, 112, 14786 CAS.
- Y. Luo, Y. Tian, A. Zhu, H. Liu and J. Zhou, J. Electroanal. Chem., 2010, 642, 109 CrossRef CAS.
- Y. Luo, H. Liu, Q. Rui and Y. Tian, Anal. Chem., 2009, 81, 3035 CrossRef CAS.
- L. Liu, N. Wang, X. Cao and L. Guo, Nano Res., 2010, 3, 369 CrossRef CAS.
- F. Hawkridge and T. Kuwana, Anal. Chem., 1973, 45, 1021 CrossRef CAS.
- A. Moghaddam, M. Ganjali, R. Dinarvand, T. Razavi, A. Saboury, A. Moosavi-Movahedi and P. Norouzi, J. Electroanal. Chem., 2008, 614, 83 CrossRef CAS.
- A. Bard and L. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 1980 Search PubMed.
- R. Nicholson, Anal. Chem., 1965, 37, 1351 CrossRef CAS.
- E. Bowden and F. Hawkridge, J. Electroanal. Chem., 1984, 161, 355 CrossRef CAS.
- W. Koppenol, C. Vroonland and R. Braams, Biochim. Biophys. Acta, Bioenerg., 1978, 503, 499 CrossRef CAS.
- G. Battistuzzi, M. Borsari, D. Dallari, I. Lancellotti and M. Sola, Eur. J. Biochem., 1996, 241, 208 CAS.
- K. Koller and F. Hawkridge, J. Electroanal. Chem., 1988, 239, 291 CrossRef CAS.
- S. Oellerich, H. Wackerbarth and P. Hildebrandt, J. Phys. Chem. B, 2002, 106, 6566 CrossRef CAS.
- D. Jokic, Z. Asfari and J. Weiss, Org. Lett., 2002, 4, 2129 CrossRef CAS.
- M. Makarska, St. Radzki and J. Legendziewicz, J. Alloys Compd., 2002, 341, 233 CrossRef CAS.
- H. Nakatani and H. Dunford, J. Phys. Chem., 1979, 83, 2662 CrossRef CAS.
- Á. Szücs and M. Noák, J. Electroanal. Chem., 1995, 383, 75 CrossRef.
- D. Chen, G. Wang and J. Li, J. Phys. Chem. C, 2007, 111, 2351 CAS.
- L. Zhang, Q. Zhang, X. Lu and J. Li, Biosens. Bioelectron., 2007, 23, 102 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |