DOI:
10.1039/C2RA01164F
(Paper)
RSC Adv., 2012,
2, 4720-4727
Hierarchical mesoporous TiO2 microspheres for the enhanced photocatalytic oxidation of sulfonamides and their mechanism†
Received
23rd November 2011
, Accepted 10th March 2012
First published on 13th April 2012
Abstract
Sulfonamides, a group of antibacterial agents, are not readily biodegradable and as a result have often been detected in wastewater effluents, rivers and lakes. This work investigated the photocatalytic degradation of sulfadimethoxine (SDM) and related sulfonamides in H2O2 containing aqueous suspensions of mesoporous TiO2 microspheres exposed to simulated solar light irradiation. The three-dimensional mesoporous TiO2 microsphere catalyst was fabricated by a facile method via a one-step solvothermal process without templates. The performance of mesoporous TiO2 microspheres exceeded the commercially available P25 TiO2 catalyst in the photocatalytic degradation of sulfonamides. Sulfadimethoxine degradation increased and reached the optimal H2O2 concentration of 5.9 mM. Particularly, the disappearance of SDM as well as the formation and decomposition of some degradation intermediates were determined by high-performance liquid chromatography-mass spectroscopy (HPLC-MS) and high-performance liquid chromatography-selective ion recording (HPLC-SIR) data modules, and possible photocatalytic degradation mechanisms were proposed. The hydroxylation and cleavage of the S–N or C–N bonds through •OH attacks on the aromatic rings and aminopyrimidine rings under simulated solar light irradiation with the assistance of H2O2 played important roles during SDM photocatalytic degradation.
1. Introduction
Sulfa drugs or sulfonamides are synthetic antimicrobial agents commonly used in human and veterinary medicine. Sulfonamides are among the most frequently detected antibiotics in surface waters.1 They are not readily biodegradable in the natural environment,2 leading to the common detection of such compounds in groundwater, secondary wastewater effluent, landfill leachate, and soils irrigated with reclaimed water.3–5 Research indicates that sulfonamides in water may be removed by a chemical oxidation processes.6–8 Photocatalytic oxidation is a promising technology because it is efficient in carbon mineralization and can utilize sunlight as an energy source.9 Titanium dioxide (TiO2) induced photocatalysis is a technology widely employed in water and wastewater treatment recently to eliminate organic compounds due to its particular optical properties, innocuity, low cost, and durability in terms of photo and chemical corrosion.10,11 Most efforts have been dedicated to the UV-light-driven photocatalytic oxidation process that typically employed TiO2. As the photocatalyst, TiO2 is a high band gap (∼3.2 eV) semiconductor material and can be used for photocatalysis by the illumination of ultraviolet (UV) light. However, UV light only accounts for a small portion (∼5%) of the solar spectrum in comparison to the visible region (∼45%). Designing new photocatalytic materials with visible light activation is therefore of great interest. To achieve this purpose, efforts have been devoted to shift the optical response of TiO2 from the UV to the visible spectral range to effectively utilize solar energy. The addition of oxidizing agents such as H2O2 has proved to be an effective approach.12–14 It is known that the chemisorption of H2O2 on the surface of TiO2 results in the formation of the yellow complex “Titanium peroxide”.15 Although the interaction between H2O2 and TiO2 is well-documented, the oxidation mechanism of organic compounds in the TiO2/H2O2 system under solar light irradiation is not well-understood.14
Significant advances have been made in the utilization of TiO2 nanoparticles in the photocatalytic degradation of organic waste on the laboratory scale; however, the obvious drawback of TiO2 nanoparticles is their small size, making separation difficult and restricting their applications on large scales. Mesoporous materials, with relatively large particles, have attracted a lot of attention because they have large surface areas (up to 2000 m2 g−1) and uniform mesopores between 2–20 nm that give them with high chemical sorption kinetics.16 Mesoporous nanocrystalline titania has proved to afford more active sites for adsorption and photocatalytic reactions, providing excellent activity for the photo-oxidation of formaldehyde and acetone mixtures in the gas phase at room temperature.17 Researchers reported the high efficiency of photo-degrading dyes in water by using mesoporous anatase TiO2 catalyst.18–20 Mesoporous TiO2 microspheres were fabricated and showed much better photoactivity than that of commercial photocatalyst P25 TiO2 (Degussa, Germany) due to its large surface area, highly crystallized mesoporous wall, and its greater number of active sites for concentrating the substrate.21–23
While several previous studies have reported photocatalytic oxidation of sulfonamides in UV, UV/TiO2, Vis/TiO2 and UV/TiO2/FeCl3 systems,7,24–27 in this paper, mesoporous TiO2 microspheres were employed to investigate their photoactivity on sulphonamide degradation. This is the first to report the photocatalysis of sulfadimethoxine (SDM) in the suspension of mesoporous TiO2 microspheres under simulated solar light irradiation. The influence of H2O2 was evaluated and a degradation mechanism was proposed. Under the optimal photoreaction conditions, the photodegradation intermediates of SDM were examined by high performance liquid chromatography-mass spectroscopy (HPLC-MS) and high-performance liquid chromatography-selective ion recording (HPLC-SIR) data module. Possible mechanisms of product formation during photocatalytic degradation are also proposed under the present experimental conditions.
2. Experimental
2.1. Materials and reagents
Sulfonamides standards with purity > 99% were purchased from Sigma-Aldrich and used as received (Table 1). Degussa P25 (anatase![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :rutile = 80:20, particle size 21 nm, BET area 50 m2 g−1) was supplied from Degussa AG. Acetonitrile and methanol (HPLC grade) were obtained from Dikma Scientific (Dikma, Beijing, China). Other chemicals used were all in analytical grade and purchased from Tianjin Chemical Reagents Company (Tianjin, China). Ultrapure water was prepared with a Milli-Q water purification system (Millipore, Bedford, MA, USA).
:rutile = 80:20, particle size 21 nm, BET area 50 m2 g−1) was supplied from Degussa AG. Acetonitrile and methanol (HPLC grade) were obtained from Dikma Scientific (Dikma, Beijing, China). Other chemicals used were all in analytical grade and purchased from Tianjin Chemical Reagents Company (Tianjin, China). Ultrapure water was prepared with a Milli-Q water purification system (Millipore, Bedford, MA, USA).
Table 1 Structure and pKa of selected sulphonamides
| Sulphonamides |
Acronym |
CAS |
Structure |
pKa1 |
pKa2 |
Ref. |
| Sulfacetamide |
SAAM |
144-80-9 |

|
1.8 |
5.4 |
28
|
| Sulfapyridine |
SPD |
144-83-2 |

|
2.74 |
8.29 |
28
|
| Sulfamethazine |
SMZ |
57-68-1 |

|
2.3 |
7.4 |
29
|
| Sulfisoxazole |
SIA |
127-69-5 |

|
1.5 ± 0.2 |
5.0 ± 0.7 |
29
|
| Sulfadimethoxine |
SDM |
122-11-2 |
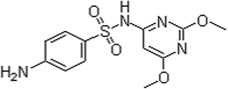
|
2.9 ± 0.5 |
6.1 ± 0.2 |
29
|
2.2 Preparation of mesoporous TiO2 microspheres
The mesoporous TiO2 microspheres were synthesized via a one-step solvothermal process without templates. An appropriate amount of NaOH was completely dissolved in 40 ml ethanol and 2 ml TiCl3 was added dropwise to the solution with vigorous stirring. The resultant transparent solution was put into a Teflon-lined autoclave vessel and maintained at 150 °C for 24 h. After autoclaving, the content was naturally cooled to room temperature and centrifuged to collect the solids that were rinsed thoroughly with distilled water and ethanol for several times. The powder was dried at 60 °C for 6 h, and then calcined at 400 °C for 2 h. This product was denoted as TiO2 3DM.
2.3 Photocatalysis experiments
Photocatalytic reaction experiments were performed in a photochemical reactor system as described previously (ESI Fig. S1†). The reaction vessel was made of quartz and was suitable to accommodate an immersion well. Simulated sunlight irradiation was provided by an 800 W xenon lamp and UV irradiation was provided by a 300 W medium-pressure mercury lamp (Institute of Electric Light Source, Beijing), which was positioned in a cylindrical quartz cold trap. The reaction solution was stirred by a magnetic stirrer at the bottom of the reactor. Two hundred milliliters of aqueous suspension containing TiO2 and sulfonamides were equilibrated in darkness for at least 30 min prior to irradiation. After collecting an initial aliquot from the equilibrated suspension in the dark, a batch reaction was initiated by placing the suspension in the collimated light path. One-milliliter aliquots of suspension were periodically collected from the irradiated batch reactor, and TiO2 was separated from the aqueous phase by centrifugation (3000 rpm, 10 min).
2.4 Analytical quantification procedures
The HPLC separation was performed using a Waters 2695 HPLC separation module (Waters, USA) equipped with a Kromasil C18 column (250 × 4.6 mm, particle size 5 μm). The column was maintained at 25 °C during the sample analysis. The mobile phase consisted of eluent A (acetonitrile) and eluent B (0.1% formic acid in ultrapure water). The flow rate was kept at 0.4 ml min−1, and the injection volume was 10 μL. The separation of antibiotics was achieved with a gradient program as follows: 0–10 min: 15–50% A; 10–16 min: 50–80% A; 16–21 min: 80% A; 21–26 min: 80–15% A; 26–30 min: 15% A. The system was re-equilibrated for 10 min between runs.
The tandem MS analyses were carried out on a Micromass Quattro triple-quadrupole mass spectrometer equipped with an electrospray ionization (ESI) source and operated in the positive ion mode. The optimal conditions for the MS system were determined as follows: source temperature 90 °C, desolvation temperature 300 °C, desolvation gas flow 500 L h−1, cone gas flow 70 L h−1 and capillary voltage 4.0 kV. Multiple responses monitoring (MRM) analysis was used to verify the five sulfonamides in the reaction solution. The optimal collision energy, cone voltage and transitions chosen for MRM were determined (ESI, Table S1†). For each analyte, quantification was based on the response of a single product ion (Table S1†).
Organic reaction intermediates were identified using liquid chromatography with tandem mass spectrometry (LC/MS/MS). MS analyses were conducted in positive mode with ESI over a mass range of 50–400 m/z, and an ion trap MS was used to separate ions with different m/z values. Selective ion recording (SIR) mode with a dwell time of 200 ms was used to acquire MS spectra and MS/MS spectra of analyte and its intermediates.
3. Results and discussion
3.1 Characterization of prepared catalysts
The mesoporous TiO2 microspheres, with or without calcination, exhibit similar characteristics, as shown on wide angle XRD patterns (Fig. 1) which can be indexed to 25.3 (101), 37.2 (004), 48.9 (200), 54.0 (105), 55.3 (211), 62.4(204) and 68.7 (112). The data are consistent with the reported values of JCPDS No. 21-1272, suggesting that anatase is the only crystalline phase in the samples. The mean crystal sizes of the mesoporous TiO2 microspheres were calculated from the breadth of the (101) XRD peaks of the anatase phase according to Scherrer formula as 9.1 and 5.0 nm, respectively for mesoporous TiO2 samples with and without calcination. The size and shape of the products were confirmed by Field-emission scanning electron microscopy (FESEM) and high resolution transmission electron microscopy (HRTEM) images (Fig. 2a, 2b). It shows that mesoporous TiO2 has rough surfaces, and the microspheres are formed by the aggregation of the nanoparticles. The TiO2 3DM has a nanoporous structure, and the pores are attributed to the interparticle spaces, and the nanoparticles are monocrystals (Fig. 2c, 2d).
 |
| | Fig. 1 XRD patterns of the catalysts: (a) TiO2 3DM calcined at 400 °C, (b) TiO2 3DM uncalcined, and (c) P25 TiO2. | |
 |
| | Fig. 2 Field-emission scanning electron microscopy (FESEM) and high resolution transmission electron microscopy images (HRTEM) of the 400 °C calcined TiO2 3DM surface: (a) (b) low magnification, (c) (d) high magnification. | |
According to the IUPAC classification, the N2 adsorption/desorption isotherms of the mesoporous TiO2 microspheres (ESI, Fig. S2†) show a typical IV pattern with hysteresis loop, which is related to the capillary condensation associated with large mesopores.30 The Barrett–Joyner–Halenda (BJH) pore size distribution profiles (Fig. S2 insert†) reveals a small hysteresis loop, implying the presence of mesopores at a high relative pressure (P/P0) range between 0.4 and 0.8.31 The formation of the mesoporous structure can be ascribed to the aggregation of TiO2 particles. Moreover, the hysteresis loops for the sample prepared with calcination at 400 °C shifts to a higher P/P0 range and the areas of the hysteresis loops become bigger than that of the uncalcined, indicating the increased mean pore size and pore volume with increasing calcination temperatures.31 The properties of TiO2 3DM are as follows: mean particle size of ca. 11.6 nm, crystallite size of ca. 9.1 nm, and specific surface area of 140.9 m2 g−1. The TiO2 3DM shows a stronger absorption in the UV-vis light region compared to that of P25 TiO2. See diffuse reflectance spectra of TiO2 3DM and P25 TiO2 for supplementary information on the absorption edges of the two catalysts (Fig. S3†). An intensive absorption band with a steep edge was observed, indicating that the apparently higher adsorption of mesoporous TiO2 microspheres might be from their reflective behavior. The enhancement of absorbance in the UV-vis region increases the number of photogenerated electrons and holes to participate in the photocatalytic reaction which will enhance the photocatalytic activity of the catalyst.32
3.2 Photodegradation of SDM and related antimicrobials in aqueous solution
When sulfonamides in reaction solutions were photo-oxidized in the presence of TiO2 3DM under simulated solar light irradiation, the substrates dramatically decreased with time and eventually disappeared after 60-min exposure. Chromatograms in Fig. S4, ESI† showed the evolution of substrates during the photo-catalytic oxidation process. The dark control showed minimal SDM adsorption to TiO2 surfaces and almost no chemical reaction occurs without sunlight (Fig. 3a). The reaction solution irradiated with simulated solar light and UV light only degraded 16.4% and 33.1% of SDM, respectively, in the 60 min exposure. In the presence of TiO2 3DM and irradiated with simulated solar light, 81.6% of SDM in the reaction solution was degraded within 60 min, a better performance than that of the commercial P25 TiO2 (Fig. 3a). The UV-irradiated TiO2 3DM catalytic oxidation completely removed the SDM and four sulfonamides analogues in the reaction solutions in 20 min (Fig. 3b). The TiO2 3DM exhibited much higher photocatalytic activity than the P25 TiO2. For the TiO2 suspension under UV irradiation, the oxidative activity of TiO2 particles is mostly attributed to the species formed through reactions initiated by the photo-generated electron (e−)-hole (h+) pairs, such as hydroxyl radicals, hydroperoxyl and other peroxyl radicals, valence-band holes in TiO2, and hydrogen peroxide.33 A photon absorbed by an organic molecule causes an electronic excitation of the molecule following the basic photochemical law of Gilbert and Baggott.34 The UV irradiation accelerated the degradation of all five sulfonamides (Fig. 3b). The photocatalytic process is initiated when a semiconductor such as TiO2 absorb photons with an energy equal or superior to their band gap, promoting the valence band electrons to the conduction band energy level. The electrons in the conduction band (eCB−) can recombine with the photo-generated vacancies (holes, hVB+) decreasing the efficiency of the photocatalytic process. TiO2 3DM showed a photoresponse in both the UV and solar light regions, accordingly, TiO2 3DM has greater speed and effectiveness for degrading sulfonamide analogues under UV irradiation, which gives higher populations of the photoexcited e− and h+ to participate in the photocatalytic reaction in this system.35
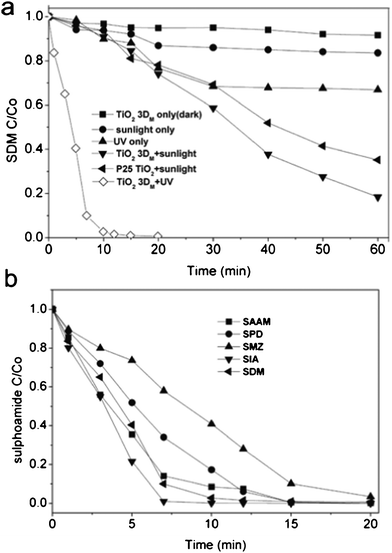 |
| | Fig. 3 (a) Photocatalytic degradation of SDM (b) UV-TiO2 3DM catalyzed photo degradation rates for five sulfonamide analogues. Reaction conditions: Co = 0.1 mM, 0.5 g L−1 of TiO2 3DM and P25 TiO2, and 25 °C | |
The photocatalytic degradation of sulfonamides as a function of the irradiation time by TiO2 3DM and P25 TiO2 was observed to follow first-order kinetic reaction:9
| |  | (1) |
where kapp represents the apparent degradation rate constant, which was determined by plotting ln(C/C0) versus reaction time t. The values of kapp for SDM in TiO2 3DM and P25 TiO2 suspensions under simulated solar light are 0.029 and 0.018 min−1, respectively, indicating the preferable photocatalytic performance of TiO2 3DM which possessed a suitable conformation of pores allowing light waves to penetrate deep inside the photocatalyst and leading to a high mobility of charge.36,37 The smaller the pore sizes, the stronger the penetration of light waves. The large specific surface area of TiO2 3DM also facilitates the absorption and utilization of solar light, which is also essential for the photocatalytic degradation.20,38 The experimental outcomes confirmed that the photocatalytic effects of TiO2 3DM were stronger than that of P25 TiO2.
The formation of the hydroxyl radicals on the surface of the mesoporous TiO2 microspheres catalysts under simulated solar light irradiation was monitored by photoluminescence (PL) with terephthalic acid as a probe molecule which can readily react with •OH to produce highly fluorescent product, 2-hydroxyterephthalic acid.39,40 The experimental procedure was similar to the photocatalytic process, except that the sulfonamides solution was replaced by the 5 × 10−4 M terephthalic acid solution in 2 × 10−3 M NaOH. The fluorescence spectra of the formed 2-hydroxyterephthalic acid were measured by a Hitachi F-4500 spectrophotometer excited at 315 nm. A higher recombination rate was observed from the higher PL intensity during the reaction process (Fig. S5, ESI†), indicating the formation of hydroxyl radicals during the photocatalytic degradation the present system.
3.3 The effect of H2O2 on the photodecomposition of sulfonamides and its degradation mechanism
Adding powerful oxidizing species such as hydrogen peroxide (H2O2) and potassium peroxydisulfate (K2S2O8) to TiO2 suspensions may increase the photo-oxidation rates.41 H2O2 plays dual roles in enhancing the semiconductor-sensitized photocatalytic degradation of organic compounds by acting as an electron scavenger to prevent the recombination of e− and h+ and as a direct source of hydroxyl radicals.26,42
The standard conditions for the photocatalytic degradation experiments involved 0.1 mM SDM in the presence of 0.5 g L−1 TiO2 3DM exposed to 30 min of simulated solar light irradiation. When H2O2 added, from 1.47 to 11.76 mM, the substrate in the reaction solution initially disappeared with amount of H2O2 added up to C = 5.88 mM, beyond which less substrate was degraded with more H2O2 added (Fig. 4a). As the SDM was almost completely eliminated at initial H2O2 concentrations of 5.88 mM, it was selected as the optimal H2O2 treatment for sequent experiments. The resulting first-order rate constants k are 0.060, 0.066, 0.089, 0.092 and 0.095 min−1 for SAAM, SPD, SMZ, SIA and SDM, respectively (Fig. S6).
 |
| | Fig. 4 Photo-catalytic oxidation of 0.1 mM SDM with presence of 0.5 g L−1 TiO2 3DM under simulated solar light for 30 min (a) H2O2 concentrations and (b) different reaction conditions with initial H2O2 concentrations of 5.88 mM. | |
Comparing outcomes of the treatment combinations, the TiO2-3DM + H2O2 without irradiation, H2O2 with irradiation only, and P25 TiO2 + H2O2 with irradiation removed 5%, 21%, and 45% of SDM in the reaction solution in 30 min of contact time, respectively (Fig. 4b). The TiO2-3DM + H2O2 with irradiation treatment however resulted in a pronounced enhancement of the reaction and SDM was almost completely removed after 30 min of contact. The H2O2 by itself was ineffective in degrading SDM. The percentages of SDM degradation employing 30 min of simulated solar light irradiation in the presence of H2O2 only, P25 TiO2 + H2O2 and TiO2-3DM + H2O2 were 21%, 45%, 99.5%, respectively (Fig. 4b). The fabricated mesoporous TiO2 microspheres were efficient at SDM removal, maybe due to their large surface area, highly crystallized mesoporous wall, and greater number of active sites for concentrating the substrate. According to the fluorescence spectral results (Fig. S5, ESI†), hydroxyl radicals were formed on the surface of the mesoporous TiO2 microsphere catalysts under simulated solar light irradiation. Additional hydroxyl radicals might be produced by the breakdown of H2O2 on the surface of TiO2 under the irradiation of solar light (eqn (2)), while a possible reaction of H2O2 with the photogenerated intermediates cannot be excluded. The enhancement of substrate degradation at H2O2 dosages from 1.5 to 5.9 mM is the result of H2O2 photolysis by solar light that generates free radicals,43 likely the dominant rate-improving mechanism in this process. H2O2 is a better electron acceptor than oxygen and may contribute to the enhanced substrate degradation in a minor way.9,44 The electrons on the conduction band of TiO2 can initiate the decomposition of H2O2 generating hydroxyl radicals, as shown in eqn (3), and forming •OH radicals via superoxide according to eqn (4).14,42 The excessive H2O2 may compete with the organic compound for the adsorption sites on the catalyst surface, resulting in a “chromatographic peaking effect” of the pollutant concentration in the solution during the initial stages of the photocatalytic process.22 Meanwhile, the excess H2O2 however also may act as a hole that scavenges the •OH and reacts with the electron-hole to form O2 or HO2• (eqn(5)). The HO2• not only serves as an exogenous active oxygen source reacting with organic compounds, but also scavenges the •OH (eqn(6)). It is imperative that the H2O2 concentration be optimal to achieve the maximum photocatalytic degradation of the substrate. The proposed photocatalysis process was illustrated in Scheme 1.
| | | H2O2 + O−2 → •OH + OH− + O2 | (4) |
| | | H2O2 + HO• → H2O + HO•2 | (5) |
 |
| | Scheme 1 Proposed photocatalysis process of the sulfonamides in aqueous suspension. | |
3.4 Degradation intermediates of SDM
The HPLC-ESI-SIR mass spectrometry chromatogram (Fig. 5) shows the potential organic intermediates 10 min after SDM in a reaction solution subjected to TiO2-3DM + H2O2 photocatalytic oxidation. Fig. S7, ESI† provided independent verification of the peaks in Fig. 5. Seven products were identified by the molecular ions and mass fragment peaks and by comparison with HPLC-MS library data (Fig. 6 and Fig. S8†). The molecular structure of SDM is displayed in Table 1; it has a [M+H]+ 311, which was eluted as peak 7 in Fig. 5 (Rt = 22.65 min, Fig. S8†). Calza et al.24 reported that in the photocatalytic degradation of SDM using P25 TiO2 powder only three intermediates were confirmed which corresponded to peak 4, peak 5, and peak 1 or 6 in Fig. 5. Two peaks holding [M+H]+ 327 (Fig. 5 peak 4, Rt = 20.43 min and peak 5, Rt = 21.91 min) present retention times shorter than the parent molecule, suggesting a proof for the occurrence of •OH radicals in the present system and the formation of a hydroxylated structure by an •OH radical attack,45 which was also confirmed by the experimental results (Fig. S5, ESI†). The intermediates holding [M+H]+ 156 (Fig. 5 peak 1, Rt = 18.61 min and peak 6, Rt = 22.56 min) were found in our experiment, following the fragmentation pathway identified by the MS/MS spectra. The intermediate occurs through the cleavage of the S–N bond, and is attributed to the species 2,5-cyclohexadiene-1-thione-4-imino-dioxide and 2,6-dimethoxy-4-aminopyrimidine (Fig. 6).
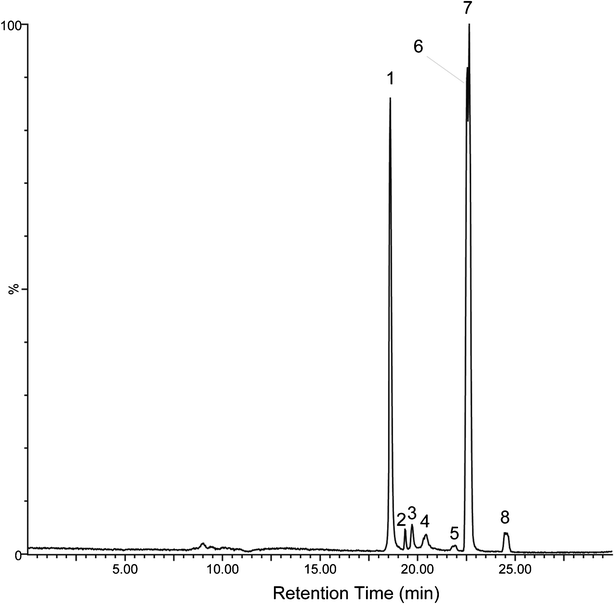 |
| | Fig. 5 HPLC-ESI-SIR mass spectrometric chromatogram showing degradation intermediates of SDM photolysis in the presence of TiO2 3DM and H2O2 and after 10 min of exposure to simulated solar light irradiation. Peaks are cross-referenced with outcomes of liquid chromatography mass spectrometry (HPLC-MS) shown in Fig. S7.† | |
 |
| | Fig. 6 Proposed products of photocatalytic degradation of SDM in the presence of mesoporous TiO2 microspheres and H2O2 exposed to simulated solar light irradiation. | |
In this study, the photocatalytic degradation of SDM produces additional products compared with the referenced literatures.24,27 One important direct pathway is through excitation of SDM to its triplet state followed by self-quenching by ground state SDM to produce the SO2 extrusion product 4-(6-imino-2,4-dimethoxypyrimidin-1(6H)-yl) aniline, which has been identified by other researchers29,46 as [M+H]+ 247 mass unit corresponding to peak 2, Rt = 19.35 min in Fig. 5. One more intermediate product with [M+H]+ 172 with the m/z lost by 140, corresponding to peak 3, Rt = 19.72 min in Fig. 5 and was identified as 4-amino-benzene sulphonamide. Its formation may be attributed to the cleavage of the C–N bond, the mechanism used to explain the formation of sulfamethoxazole degradation intermediates by TiO2 and TiO2/FeCl3 in aquatic environments.25 The last compound with the [M+H]+ 341 mass unit is somewhat suspect as it would be formed from O-addition only to the phenyl ring or addition to both rings (peak 8, Rt = 24.51 min in Fig. 5).46 The possible bisoxidation intermediate (SDM+32) in the experimental system was identified by an electrospray ionization (ESI) source and in the positive ion mode.
4. Conclusions
In this work, pure ordered mesoporous TiO2 microspheres were successfully synthesized via a one-step solvothermal process without templates. The obtained TiO2 3DM are highly active for the photodegradation of sulfadimethoxine and related sulfonamides antimicrobial agents. Due to their specific properties, these products have advantages over commercial P25 TiO2 for photocatalytic activities. Our research has shown that SDM can undergo significant photocatalytic degradation by mesoporous TiO2 microspheres with the assistance of H2O2 under simulated solar light irradiation, suggesting that these products will provide the possibility of being employed in the cleaning up of industrial pollutants. The seven intermediates of SDM identified by LC/ESI-MS and LC/ESI-SIR in this study have not been simultaneously proposed in previous research. However, although photocatalysis is efficient in degrading SDM, some of its photoproducts may exhibit residual antimicrobial activity which is the focus of the future research.
Acknowledgements
This work was supported by National Natural Sciences Foundation of China (20977051).
References
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS.
- F. Ingerslev and B. Halling-Sørensen, Environ. Toxicol. Chem., 2000, 19, 2467–2473 CrossRef CAS.
- X.-S. Miao, F. Bishay, M. Chen and C. D. Metcalfe, Environ. Sci. Technol., 2004, 38, 3533–3541 CrossRef CAS.
- J. V. Holm, K. Ruegge, P. L. Bjerg and T. H. Christensen, Environ. Sci. Technol., 1995, 29, 1415–1420 CrossRef CAS.
- J. A. Pedersen, M. Soliman and I. H. Suffet, J. Agric. Food Chem., 2005, 53, 1625–1632 CrossRef CAS.
- V. K. Sharma, S. K. Mishra and N. Nesnas, Environ. Sci. Technol., 2006, 40, 7222–7227 CrossRef CAS.
- L. Hu, P. M. Flanders, P. L. Miller and T. J. Strathmann, Water Res., 2007, 41, 2612–2626 CrossRef CAS.
- M. C. Dodd and C.-H. Huang, Environ. Sci. Technol., 2004, 38, 5607–5615 CrossRef CAS.
- D. F. Ollis, E. Pelizzetti and N. Serpone, Environ. Sci. Technol., 1991, 25, 1522–1529 CrossRef CAS.
- V. Augugliaro, V. Loddo, L. Palmisano and M. Schiavello, J. Catal., 1995, 153, 32–40 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- T. Ohno, Y. Masaki, S. Hirayama and M. Matsumura, J. Catal., 2001, 204, 163–168 CrossRef CAS.
- X. Li, C. Chen and J. Zhao, Langmuir, 2001, 17, 4118–4122 CrossRef CAS.
- Y. F. Rao and W. Chu, Environ. Sci. Technol., 2009, 43, 6183–6189 CrossRef CAS.
- A. H. Boonstra and C. A. H. A. Mutsaers, J. Phys. Chem., 1975, 79, 1940–1943 CrossRef CAS.
- K. Hanna, I. Beurroies, R. Denoyel, D. Desplantier-Giscard, A. Galarneau and F. Di Renzo, J. Colloid Interface Sci., 2002, 252, 276–283 CrossRef CAS.
- J. G. Yu, M. H. Zhou, B. Cheng, H. G. Yu and X. J. Zhao, J. Mol. Catal. A: Chem., 2005, 227, 75–80 CrossRef CAS.
- D. S. Kim and S. Y. Kwak, Appl. Catal., A, 2007, 323, 110–118 CrossRef CAS.
- Z. Liu, H. Bai and D. Sun, Appl. Catal., B, 2011, 104, 234–238 CrossRef CAS.
- H. Einaga, T. Ibusuki and S. Futamura, Environ. Sci. Technol., 2003, 38, 285–289 CrossRef.
- C. Guo, M. Ge, L. Liu, G. Gao, Y. Feng and Y. Wang, Environ. Sci. Technol., 2010, 44, 419–425 CrossRef CAS.
- G. Kawamura, M. Murakami, T. Okuno, H. Muto and A. Matsuda, RSC Adv., 2011, 1, 584–587 RSC.
- F. Bosc, D. Edwards, N. Keller, V. Keller and A. Ayral, Thin Solid Films, 2006, 495, 272–279 CrossRef CAS.
- P. Calza, C. Medana, M. Pazzi, C. Baiocchi and E. Pelizzetti, Appl. Catal., B, 2004, 53, 63–69 CrossRef CAS.
- W. Baran, E. Adamek, A. Sobczak and A. Makowski, Appl. Catal., B, 2009, 90, 516–525 CrossRef CAS.
- S. Kaniou, K. Pitarakis, I. Barlagianni and I. Poulios, Chemosphere, 2005, 60, 372–380 CrossRef CAS.
- Y. Lester, I. Gozlan, D. Avisar and H. Mamane, Water Sci. Technol., 2008, 58, 1147–1154 CrossRef CAS.
- H. Niu, Y. Cai, Y. Shi, F. Wei, J. Liu, S. Mou and G. Jiang, Anal. Chim. Acta, 2007, 594, 81–92 CrossRef CAS.
- A. L. Boreen, W. A. Arnold and K. McNeill, Environ. Sci. Technol., 2005, 39, 3630–3638 CrossRef CAS.
- A. A. Ismail, D. W. Bahnemann, I. Bannat and M. Wark, J. Phys. Chem. C, 2009, 113, 7429–7435 CAS.
- J. Yu, L. Zhang, B. Cheng and Y. Su, J. Phys. Chem. C, 2007, 111, 10582–10589 CAS.
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Mater. Sci. Eng., B, 2005, 117, 67–75 CrossRef.
- K. Dai, T. Peng, H. Chen, J. Liu and L. Zan, Environ. Sci. Technol., 2009, 43, 1540–1545 CrossRef CAS.
-
E. Gilbert and J. Baggott, Essentials of Molecular Photochemistry, Blackwell Scientific Publications, Oxford, 1991 Search PubMed.
- R. A. Palominos, M. A. Mondaca, A. Giraldo, G. Peñuela, M. Pérez-Moya and H. D. Mansilla, Catal. Today, 2009, 144, 100–105 CrossRef CAS.
- X. Wang, J. C. Yu, C. Ho, Y. Hou and X. Fu, Langmuir, 2005, 21, 2552–2559 CrossRef CAS.
- L. Z. Zhang and J. C. Yu, Chem. Commun., 2003, 2078–2079 RSC.
- H. Jean-Marie, Catal. Today, 1999, 53, 115–129 CrossRef.
- A. C. Pradhan, K. Parida and B. Nanda, Dalton Trans., 2011, 40, 7348–7356 RSC.
- X. Yu, S. Liu and J. Yu, Appl. Catal., B, 2011, 104, 12–20 CrossRef CAS.
- S. Malato, J. Blanco, M. I. Maldonado, P. Fernández-Ibáñez and A. Campos, Appl. Catal., B, 2000, 28, 163–174 CrossRef CAS.
- C. C. Wong and W. Chu, Environ. Sci. Technol., 2003, 37, 2310–2316 CrossRef CAS.
- W. Chu and W. K. Choy, Water Res., 2002, 36, 2525–2532 CrossRef CAS.
- I. Ilisz, K. Föglein and A. Dombi, J. Mol. Catal. A: Chem., 1998, 135, 55–61 CrossRef CAS.
- S. Gazi and R. Ananthakrishnan, Curr. Anal. Chem., 2012, 8, 143–149 CrossRef CAS.
- J. J. Guerard, Y.-P. Chin, H. Mash and C. M. Hadad, Environ. Sci. Technol., 2009, 43, 8587–8592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures: (S1) The experimental apparatus, (S2) N2 adsorption/desorption isotherms of the mesoporous TiO2 microspheres, (S3) UV-vis reflection spectra of the catalysts, (S4) Mass chromatograms of five sulfonamides solutions after different simulated solar light irradiation time with TiO2 3DM, (S5) Temporal fluorescence spectral, (S6) Kinetic linear simulation curve of five sulfonamides photocatalytic degradation by mesoporous TiO2 microspheres under simulated solar light irradiation with the assistance of H2O2, (S7) Liquid chromatography mass spectrometry (LC-MS) chromatogram for SDM photocatalysis, (S8) Mass spectra corresponding proposed structures of six main peaks and SDM in TIC. Table: (S1) Analyte acronyms, molecular weights, ions monitored for LC–MS/MS, and conditions of cone voltages and collision energies. See DOI: 10.1039/c2ra01164f |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.