The influence of ionic liquid on phase separation of poly(N-isopropylacrylamide) aqueous solution†
Zhangwei
Wang
and
Peiyi
Wu
*
The State Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, and Laboratory of Advanced Materials, Fudan University, Shanghai 200433, China. E-mail: peiyiwu@fudan.edu.cn
First published on 6th July 2012
Abstract
The role of ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]), on the phase transition behavior of concentrated PNIPAM solutions was investigated by FTIR spectroscopy in combination with two-dimensional correlation spectroscopy (2Dcos) and the perturbation correlation moving window (PCMW) technique for the first time. At low IL concentrations, the Tp of the PNIPAM solution decreases with increases in the IL concentration, due to the destabilization of the hydrated macromolecule structure via preferential interactions between IL and water molecules. However, at higher IL concentrations, unexpectedly, the phase transition behavior disappears. This has been attributed to the formation of a stable interaction network via intra- and intermolecular hydrogen bonding. Furthermore, two changes can be observed in the ν(C–H) region for the sample with 0.4 mol L−1 [Bmim][BF4]. The first change is related to the phase separation of the PNIPAM solution, while the second step is attributed to an IL–D2O association, which takes part in the globule construction, probably interacting with hydrophilic groups of PNIPAM. Thus, the role of ILs on the phase behavior of PNIPAM is embodied in two opposite aspects, the “destroyer” and the “constructer”.
1 Introduction
Poly(N-isopropylacrylamide) (PNIPAM), as the most representative thermo-responsive water-soluble polymer, is of great interest for applications in drug release, surface modification peptides separation, etc.1–3 Aqueous PNIPAM solutions undergo a reversible coil-to-globule transition upon heating above the lower critical solution temperature (LCST ∼32 °C), which is related to the dehydration of the hydrophobic groups and changes to hydrogen bonds containing the hydrophilic groups.4,5 Besides the different environmental variations such as temperature, pH and pressure,4,6,7 the addition of substances such as cosolvents,8–12 surfactants,13–18 salts,19–21 even ionic liquids22 also influences the phase behavior of PNIPAM in aqueous mediums, particularly ionic liquids, which have attracted much attention.Ionic liquids (ILs) are recognized as a “green” (environmentally friendly) alternative to traditional inorganic and organic solvents due to their specific properties23–25 (negligible vapor pressures, high thermal stabilities, wide electrochemical window and excellent ionic conductivities, etc.). The properties can be tuned through combinations of anions and cations, with numerous ion pairs possible. Furthermore, via the combination of polymer and IL, many previous works have shed light on their potential uses in electrochemical fields, capacitors, fuel cell electrolytes, and other fields of value.25,26 However, only a few studies have been reported that refer to the influence of IL on the PNIPAM phase behavior.
As mentioned before, PNIPAM exhibits LCST behavior in aqueous solutions, whereas an upper critical solution temperature (UCST) is observed for ILs. The UCST is strongly affected by the concentration and molecular weight of PNIPAM.25,27,28 Moreover, fluorescence, viscometric, and dynamic light scattering (DLS) techniques have been applied to investigate the effect of the imidazolium based IL, 1-benzyl-3-methylimidazolium tetrafluoroborate ([Bzmim] [BF4]) on the LCST of PNIPAM in aqueous solution at four different [Bzmim][BF4] concentrations.22 It was observed that the LCST of aqueous PNIPAM solutions decreases with increasing IL concentration. It is proposed that this is mainly due to rupture of the hydrogen bonding between polymer and water molecules, and the destabilization of the hydrate macromolecule structure. Additionally, this research investigates the physical characteristics of aqueous PNIPAM solutions, as the interactions between polymers, water molecules, and ILs, especially the variation of microstructure during the temperature or IL concentration elevation process, have been rarely studied.
In the present work, a simple IL, 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]), is introduced as an additive to aqueous PNIPAM solutions. Under the variation of IL concentration and temperature, the micro-structures and dynamics involving the interactions between polymers, water and ILs are investigated by Fourier transform infrared spectroscopy (FTIR). FTIR is an effective method to detect molecular interactions, especially for hydrogen bonding interactions. To improve the spectral resolution by capturing subtle information that is not obvious or overlapped in 1D FTIR spectra, two-dimensional correlation spectroscopy (2Dcos)29,30 in combination with the perturbation correlation moving window (PCMW)31,32 technique was used to obtain information about molecular motions or conformational changes. For aqueous PNIPAM solutions, the phase separation mechanism has previously been studied by FTIR and 2DIR,33–35 as well as for the PNIPAM in water/methanol mixture.36 In our work, it is notable that this is the first time 2DIR and PCMW are employed to further investigate the role of ILs on the phase separation of aqueous PNIPAM solution. Wherein, the great influence of IL dynamics on the phase behavior of aqueous PNIPAM solution can be further investigated.
2 Experimental
2.1 Materials and preparation
N-isopropylacylamide (NIPAM) monomers were purchased from Tokyo Kasei Kogyo Co. (Tokyo, Japan) and recrystallized from cyclohexane before use. PNIPAM was synthesized by free-radical polymerization initiated by azobis(isobutyronitrile) (AIBN) with tetrahydrofuran (THF) as the solvent. The reaction was carried out at 70 °C for 12 h under a nitrogen atmosphere. After precipitation by diethyl ether, the product was further vacuum-dried for 24 h. The molecular weight of PNIPAM, Mw 1.1 × 104, and polydispersity index, Mw/Mn 1.6, were measured by a Voyager DE-STR matrix-assisted laser desorption/ionization time-of-flight (MALDI TOF MS) mass spectrometer equipped with a 337 nm nitrogen laser. The ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) was purchased from Aladdin Corporation (>97%). To reduce the water and volatile compounds content to negligible values, the ionic liquid was vacuum-dried at 70 °C, for a minimum of 48 h, and the water content was estimated to be less than 100 ppm by a Karl Fischer titration. Fig. 1 shows the chemical structures of the PNIPAM and [Bmim][BF4].![Chemical structure of PNIPAM (a) and ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) (b).](/image/article/2012/RA/c2ra01349e/c2ra01349e-f1.gif) | ||
| Fig. 1 Chemical structure of PNIPAM (a) and ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) (b). | ||
D2O was purchased from Cambridge Isotope Laboratories Inc. (D-99.9%). For DSC and FTIR measurements, the concentration of PNIPAM was fixed at 5% (w/v), with 0–2.0 mol L−1 [Bmim][BF4] injected. All solutions with [Bmim][BF4] in D2O were placed at 4 °C for a week before DSC and FTIR measurements to ensure complete deuteration of all the N–H protons.
2.2 Instruments and measurements
Calorimetric measurements were performed on a Mettler-Toledo differential scanning calorimeter thermal analyzer with a rate of 10 °C min−1 during the heating and the detailed experimental method is shown in the ESI.† The PNIPAM solutions for FTIR measurements were prepared in a transmission cell sealing the liquid between two pieces of microscope ZnS windows, which have no absorption bands in the MIR region. The FTIR spectra were recorded with a 4 cm−1 spectral resolution on a Nicolet Nexus 470 spectrometer equipped with a DTGS detector by signal-averaging 32 scans. Temperature-dependent spectra were collected between 25 and 45 °C in intervals of 1.0 °C with an accuracy of 0.1 °C. The baseline-corrected processing was by the Omnic, ver. 6.1a.2.3 Investigation methods
3 Results and discussion
3.1 Influence of IL concentration on aqueous PNIPAM solution
![DSC curves of 5% (w/v) PNIPAM/D2O solutions with different concentrations of [Bmim][BF4] (0–2.0 mol L−1), respectively, at temperature variation rate of 10 °C min−1.](/image/article/2012/RA/c2ra01349e/c2ra01349e-f2.gif) | ||
| Fig. 2 DSC curves of 5% (w/v) PNIPAM/D2O solutions with different concentrations of [Bmim][BF4] (0–2.0 mol L−1), respectively, at temperature variation rate of 10 °C min−1. | ||
Interestingly, at a higher IL concentration, ca. 1.5 mol L−1, the endothermic peak disappears, that is, the phase transition of the PNIPAM solution was not observed. These results reveal that the addition of IL has a great influence on the phase transition of aqueous PNIPAM solutions. At the low IL concentrations, the Tp of PNIPAM solutions decreases with increasing IL concentrations; at higher IL concentrations, the phase transition behavior disappears. The different response to the variation in the IL concentration might be associated with the different formation of interactions among the ternary system.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band of PNIPAM. All the samples with 5% (w/v) PNIPAM, 0–2.0 mol L−1 [Bmim][BF4] were measured by FTIR spectroscopy at 25 °C. Two spectral regions are focused on here, the C–H stretching band (3020–2840 cm−1), and the amide I (CO hydrogen bonding) (1680–1588 cm−1). By analyzing these regions, information on the molecular motion of chemical groups affected by the variation of IL concentrations could be obtained. For clarity, the second derivative spectra of the two regions are also given to enhance the spectra resolution to further investigate the slight change induced by the different IL concentration. In Fig. 3, the absorption bands of the C–H and CO groups of the samples with 0, 0.4, 1.0, 1.5, 2.0 mol L−1 [Bmim][BF4] at 25 °C are given as examples, as well as the corresponding second derivative spectra.
O) band of PNIPAM. All the samples with 5% (w/v) PNIPAM, 0–2.0 mol L−1 [Bmim][BF4] were measured by FTIR spectroscopy at 25 °C. Two spectral regions are focused on here, the C–H stretching band (3020–2840 cm−1), and the amide I (CO hydrogen bonding) (1680–1588 cm−1). By analyzing these regions, information on the molecular motion of chemical groups affected by the variation of IL concentrations could be obtained. For clarity, the second derivative spectra of the two regions are also given to enhance the spectra resolution to further investigate the slight change induced by the different IL concentration. In Fig. 3, the absorption bands of the C–H and CO groups of the samples with 0, 0.4, 1.0, 1.5, 2.0 mol L−1 [Bmim][BF4] at 25 °C are given as examples, as well as the corresponding second derivative spectra.
![FTIR and corresponding second derivative spectra of the ν(C–H) region (a), and ν(CO) region (b) of 5% (w/v) PNIPAM in D2O with different [Bmim][BF4] concentrations (0, 0.4, 1.0, 1.5, 2.0 mol L−1) at 25 °C.](/image/article/2012/RA/c2ra01349e/c2ra01349e-f3.gif) | ||
| Fig. 3 FTIR and corresponding second derivative spectra of the ν(C–H) region (a), and ν(CO) region (b) of 5% (w/v) PNIPAM in D2O with different [Bmim][BF4] concentrations (0, 0.4, 1.0, 1.5, 2.0 mol L−1) at 25 °C. | ||
Fig. 3a shows the FTIR and corresponding second derivative spectra of the C–H stretching region, where the νas(CH3) and νas(CH2) bands centered around 2982 and 2940 cm−1 are clear. It should be noted that the νas(CH3) band shifts significantly to lower wavenumbers with the increase in the IL concentration, while the band of νas(CH2) shifts only slightly to higher wavenumber with the addition of IL. The overlap of the C–H groups of PNIPAM with the alkyl side chain of the imidazole ring enhanced the difficulties in the FTIR analysis, thus, we have used IL/D2O solutions with 0–2.0 mol L−1 [Bmim][BF4], respectively. It is unexpected that the bands of νas(CH3) and νas(CH2) appear at almost the same positions in the spectra of the mixtures with different IL concentrations, as this implies that the band shifts are mainly ascribed to the motion of PNIPAM chemical groups. It is well-known that the red shift of the C–H groups reveals the dehydration of the CH3 and CH2 groups, and the more water molecules surround the hydrophobic groups, the higher the wavenumber of the C–H stretching vibration.37 Therefore, the red shift of νas(CH3) may arise from the difference in the degree of PNIPAM side-chain collapse, that is, the variation of IL concentration has a distinct effect on the hydrated methyl groups. The more IL in the PNIPAM solution, the fewer water molecules are around the CH3 groups, which results in the destabilization of the hydrated macromolecule structure. Through IR and 2D-IR, Lendl38 discovered that water could interact with BF4− in ionic liquids by three forms of hydrogen bonds: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (BF4−⋯water), 1:2 (BF4−⋯water⋯BF4−), and a cyclic water dimer interacting with BF4−. Using simulation methods, it was found that water could interact with C2–H, C4–H, and C5–H of the cation through hydrogen bonds.39,40 Thus, the water molecules preferentially interact with [Bmim][BF4] compared with the hydrophobic CH3 groups, leading to the weakness of the hydration effect between the CH3 groups and the D2O molecules in the solution. The phase transition temperature of aqueous PNIPAM solution decreases due to the hydrophobic collapse and aggregation of polymer chains. Moreover, for the aqueous PNIPAM solutions without IL, the νas(CH2) band is located at the lower wavenumber, whereas, the variation in the IL concentration does not cause obvious changes to the CH2 groups. It is presumed that the response to IL variation for CH3 groups is different from that of CH2 groups; the latter is less sensitive, because of the limited motion space for the CH2 group on the main chains, while the CH3 groups on the PNIPAM side chains are flexible.
:1 (BF4−⋯water), 1:2 (BF4−⋯water⋯BF4−), and a cyclic water dimer interacting with BF4−. Using simulation methods, it was found that water could interact with C2–H, C4–H, and C5–H of the cation through hydrogen bonds.39,40 Thus, the water molecules preferentially interact with [Bmim][BF4] compared with the hydrophobic CH3 groups, leading to the weakness of the hydration effect between the CH3 groups and the D2O molecules in the solution. The phase transition temperature of aqueous PNIPAM solution decreases due to the hydrophobic collapse and aggregation of polymer chains. Moreover, for the aqueous PNIPAM solutions without IL, the νas(CH2) band is located at the lower wavenumber, whereas, the variation in the IL concentration does not cause obvious changes to the CH2 groups. It is presumed that the response to IL variation for CH3 groups is different from that of CH2 groups; the latter is less sensitive, because of the limited motion space for the CH2 group on the main chains, while the CH3 groups on the PNIPAM side chains are flexible.
It is especially worth noting that the FTIR second derivative spectra of 5% (w/v) PNIPAM in D2O with 0.4 mol L−1 shows two peaks centered around 2980 and 2971 cm−1. These peaks can be attributed to the vibration of the CH3 groups interacting with more or fewer D2O molecules, respectively.33 The two types of hydration interaction testifies the existence of an intermediate state, implying that the dehydration of the side-chain CH3 groups is a gradual change, not a sharp one in the IL-elevation process. At a certain IL concentration, ca. 1.5 mol L−1, represented in Fig. 3a, no obvious peak shift of the CH3 groups is observed, probably due to the formation of intra- and intermolecular hydrogen bonding, namely, stable structures are present between PNIPAM, D2O, and [Bmim][BF4]. This is consistent with the DSC results in Fig. 2, at IL concentrations above 1.5 mol L−1, the endothermic peak disappears, as well as the phase transition behavior.
The IL concentration dependence of the FTIR and corresponding second derivative spectra in the range of 1680–1588 cm−1 originating from the CO groups are shown in Fig. 3b. A slight difference in 1DIR but obvious differences in the second derivative spectra can be observed. With the addition of IL, there is a blue shift concerning the band of 1623 cm−1 assigned to the hydrogen bond of CO⋯D–O–D, revealing the weakness of the hydrogen bonding between CO groups and water molecules due to the addition of hydrophilic IL. Furthermore, it is noted that a new band at 1645 cm−1 appears when the IL concentration is at approximately 1.5 mol L−1, which should be assigned to the hydrogen bond of CO⋯D–N.41,42 This evidence confirms that intermolecular hydrogen bonds (CO hydrogen bonded with N–D) form at higher IL concentrations, in accord with our supposition that a sufficient amount of IL can lead to the formation of a stable hydrogen bonded network between the polymers, water molecules, and ILs.
It is mentioned above that ILs can modify the low-temperature state of the polymer in D2O. To further investigate the role of IL dynamics on the phase behavior of aqueous PNIPAM solution, we studied the high-temperature state of the polymer in D2O. Infrared spectra in the ν(CO) region of 5% (w/v) PNIPAM solutions with various concentrations of [Bmim][BF4] at 45 °C were measured by FTIR spectroscopy. All spectra of the amide I region could be curve-fitted simultaneously over the whole concentration range, always with two bands in the IL-elevation process. The decomposed spectra IL concentrations ranging from 0–2.0 mol L−1 are given as examples in Fig. 4a. As the bands of 1623 and 1645 cm−1 are assigned to the hydrogen bonds of CO⋯D–O–D and CO⋯D–N, respectively, it can be observed that the intensity of the two bands change with the different IL concentration. For clarity, the ratio of the peak area of the CO⋯N–D hydrogen bond related to that of the carbonyl hydrogen bonds was determined, and quantitative analysis of the changes in the peak area are shown in Fig. 4b. An interesting changing trend is found, which is probably relevant to the variations in the IL concentration. There are two main regions during the IL-elevation process: the increasing one at 0–1.0 mol L−1, and the decreasing one at 1.0–2.0 mol L−1. In the former region, at the low IL concentration, the relative areas of the CO⋯D–N hydrogen bonding goes through a slight increase with no obvious stage change, then, increases gradually with the increase of IL concentration up to 1.0 mol L−1. Below the LCST, the hydration interaction of the hydrophobic groups is destroyed by the addition of IL, together with the weakness of the CO⋯D–O–D hydrogen bonding. Thus, above the LCST, the collapse and aggregation of the macromolecules take place much more easily. The higher the IL concentration is, the more the relative component of the CO⋯D–N hydrogen bonding is, as long as the concentration of PNIPAM and the temperature are consistently maintained. In the latter region of 1.0–2.0 mol L−1, the relative area of the CO⋯D–N sharply decreases, then, up to a IL concentration of ca. 1.5 mol L−1, experiences only a gentle change. According to the previous analysis, with the higher IL concentration, the phase behavior of the PNIPAM solution disappears because of the establishment of a stable hydrogen bonded structure, which leads to the evident decrease of the relative area of CO⋯D–N. Moreover, with excess IL, the small change observed ultimately, mainly arises from more intermolecular hydrogen bonds of CO⋯D–N at a low-temperature state, and is not attributed to the thermo-response, which is referred in Fig. 3b. As mentioned above, it is interesting that the addition of IL can modify phase transition behavior of the aqueous PNIPAM solution via the variation of the IL concentration. At low IL concentration, the Tp of PNIPAM solution decreases by weakening the hydration effect of the CH3 and CO groups, while, at higher IL concentration, phase transition behavior disappears due to the formation of a stable hydrogen bonded network via intra- and intermolecular interactions in the solution.
![Deconvoluted infrared spectra of the ν(CO) region (a) and relative areas of 1645 cm−1 component (the CO ⋯ D–N hydrogen bonding) are plotted against the area of the carbonyl hydrogen bonding (b) of 5% (w/v) PNIPAM/D2O solutions with various [Bmim][BF4] concentrations (from 0 to 2.0 mol L−1) at 45 °C.](/image/article/2012/RA/c2ra01349e/c2ra01349e-f4.gif) | ||
| Fig. 4 Deconvoluted infrared spectra of the ν(CO) region (a) and relative areas of 1645 cm−1 component (the CO ⋯ D–N hydrogen bonding) are plotted against the area of the carbonyl hydrogen bonding (b) of 5% (w/v) PNIPAM/D2O solutions with various [Bmim][BF4] concentrations (from 0 to 2.0 mol L−1) at 45 °C. | ||
3.2 Temperature dependence of aqueous PNIPAM solutions with different IL concentrations
O stretching, 1680–1588 cm−1), as presented in Fig. 5 and 7, respectively, which can provide information on the molecular motion of chemical groups within the variation of IL concentration and temperature.
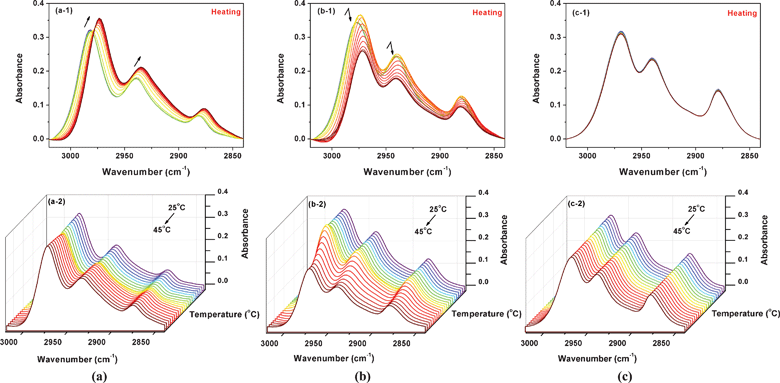 | ||
| Fig. 5 During heating from 25 to 45 °C, temperature-dependent FTIR spectra in the ν(C–H) region (a) of 5% (w/v) PNIPAM/D2O solutions with 0 mol L−1 IL, (a-1) 2D spectra and (a-2) 3D spectra; (b) of 5% (w/v) PNIPAM/D2O solutions with 0.4 mol L−1 IL, (b-1) 2D spectra and (b-2) 3D spectra; (c) of 5% (w/v) PNIPAM/D2O solutions with 1.5 mol L−1 IL, (c-1) 2D spectra and (c-2) 3D spectra. | ||
Fig. 5 shows FTIR spectra in the ν(C–H) region of 5% (w/v) PNIPAM D2O solutions with 0, 0.4, 1.5 mol L−1 IL, for clarity, the corresponding 3D spectra are also shown. It is well-known that the peak shifts during heating suggest the dehydration of the CH3 and CH2 groups, so we have focused on the spectral changes vas(CH3) around 2982 cm−1 and vas(CH2) around 2940 cm−1. For the sample without IL (Fig. 5a), the bands of vas(CH3) and vas(CH2) both shift to lower wavenumbers, which reveals dehydration of the C–H groups. As a comparison, an interesting spectral change trend is found for the sample with 0.4 mol L−1 IL (Fig. 5b), probably resulting from the addition of IL. At the beginning of heating, there is a red shift of the bands vas(CH3) and vas(CH2), together with an intensity increase. The intensities of the two peaks then experience an obvious decrease, and ultimately remain stable. Furthermore, as the IL concentration increases up to 1.5 mol L−1 (Fig. 5c), unexpectedly, both vas(CH3) and vas(CH2) experience only a slight decrease with the increase of temperature, which is probably a response to the thermal perturbation, and is not related to dehydration of the C–H groups. This observation indicates that the solutions with higher IL concentrations are not more sensitive to the temperature, which is consistent with the supposition of the existence of stable hydrogen bonded network.
In Fig. 6, the band shifts of vas(CH3) have been monitored during heating , to further explain the interesting phenomena in FTIR spectra dependent upon the variation of IL concentration. As mentioned in Fig. 3a, at 25 °C, the vas(CH3) peak shifts to lower wavenumber due to the weakness of the hydration effect by the addition of IL, that is, the more the IL is, the lower the wavenumber is, which is also illuminated in the Fig. 6. Compared with the sample without IL, during heating of the sample with 0.4 mol L−1 IL, the vas(CH3) band changes less sharply around Tp, and has a slight increase after the completion of phase transition. The evidence confirms that the existence of IL influences the hydration of CH3 groups and introduces a new microstructure, which is less sensitive to the temperature variation. Moreover, the red shift around the LCST manifests the dehydration of the side-chain CH3 groups during heating, while the final mild blue shift probably testifies the reconstruction of the interaction between CH3 groups and fewer D2O molecules originating from the dissociation of IL⋯D–O–D. Nevertheless, as the IL concentration exceeds a certain content, the peak position of vas(CH3) appears to have no significant response to the temperature variation, which implies the hydration of CH3 groups barely occurs, and the equilibrium state is achieved.
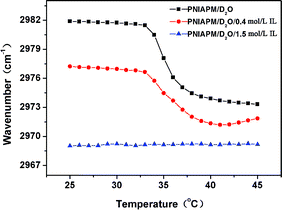 | ||
| Fig. 6 Temperature dependent peak position of vas(CH3) in the FTIR spectra of 5% (w/v) PNIPAM/D2O solutions with 0, 0.4, and 1.5 mol L−1 IL during heating from 25 to 45 °C. | ||
Peaks in the range of 1680–1588 cm−1 originating from CO stretching vibrations of 5% (w/v) PNIPAM/D2O solutions with 0, 0.4, 1.5 mol L−1 IL are shown in Fig. 7. Similarly, the spectral changing trends are different, according to the variation of IL concentration. As the bands of 1623 and 1645 cm−1 are assigned to the hydrogen bonds of CO⋯D–O–D and CO⋯D–N, respectively, it can be summarized as follows: for every sample during heating, the intensity of the band at 1623 cm−1 decreases while the intensity of the 1645 cm−1 band increases. As the concentration of IL is low, the band of 1623 cm−1 assigned to the hydrogen bonds of CO⋯D–O–D goes through a sharper intensity decrease. As the concentration is higher, this band only has a little reduction, namely the CO groups stably interact with the D2O or the IL–D2O association. A further investigation of the different change related to the phase behavior in the region will be discussed in the following PCMW and 2Dcos analysis.
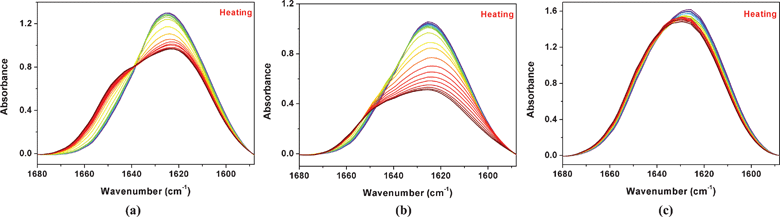 | ||
| Fig. 7 During heating from 25 to 45 °C, temperature-dependent FTIR spectra in the ν(CO) region of 5% (w/v) PNIPAM/D2O solutions with 0 (a), 0.4 (b), and 1.5 (c) mol L−1 IL, respectively. | ||
Two types of spectra (synchronous and asynchronous) can be generated by PCMW. The rules of PCMW are as follows: with the perturbation increment, a positive synchronous correlation indicates an increase in the spectral intensities, while a negative correlation indicates the decrease. Positive asynchronous correlation corresponds to a convex spectral intensity variation while a negative correlation corresponds to a concave variation. In this article, only the synchronous spectra of PCMW are presented in Fig. 8, for the determination of the phase behavior of PNIPAM/D2O/IL solutions during heating.
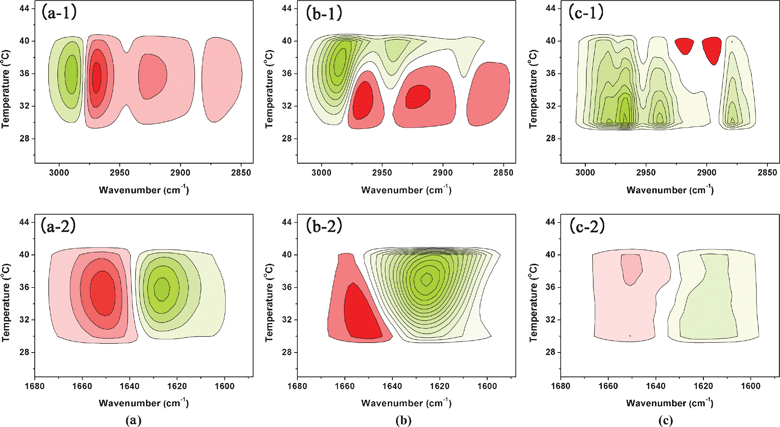 | ||
| Fig. 8 PCMW synchronous of ν(C–H) and ν(CO) region generated from the spectra of 5% (w/v) PNIPAM/D2O solutions with 0 (a), 0.4 (b), and 1.5 (c) mol L−1 IL between 25 and 45 °C. Here, warm colors (red) are defined as positive intensities, while cool colors (green) indicate negative ones. | ||
According to Morita's PCMW rule, PCMW synchronous spectra are very helpful to find transition points. Thus for C–H groups of sample with 0 mol L−1 IL (Fig. 8a-1), Tp around 35 °C, and for sample with 0.4 mol L−1 IL (Fig. 8b-1), two transition points are observed: one around 33 °C, resulting from the coil-to-globule transition; while the other is around 37 °C, and is probably related to the reduction of C–H intensities by [Bmim][BF4]. This can be further confirmed by the following 2D analysis. Meanwhile, for the sample with 1.5 mol L−1 IL (Fig. 8c-1), only one transition point occurs around 30 °C. On the basis of the DSC and 1DIR analysis, we tentatively infer that this transition point is likely caused by the motion of C–H groups with the response to the thermal perturbation, and does not refer to phase transition behavior.
It is worth noting that the change of PNIPAM CO groups can also be monitored in a PCMW synchronous map during heating, focusing on the N–D⋯OC hydrogen bonds. In Fig. 8a-2, 8b-2, and 8c-2, the intensity of CO⋯D–N hydrogen bonds around 1645 cm−1 all increase with the elevation of temperature, whereas CO⋯D–N hydrogen bonds in Fig. 8b-2 have an earlier response, Tp around 32 °C compared to that around 35 °C in Fig. 8a-2. The addition of IL leads to a reduction of the phase transition temperature. Additionally, as the IL increases up to 1.5 mol L−1 (Fig. 8c-2), the signal of the ν(CO) region in the PCMW spectra is very low, which implies there is no critical transition point of the sample. This might be ascribed to the slight motion of the CO groups during heating. These observations show that the C–H groups, as well as CO groups, have different responses to the various amounts of IL during heating, in accordance with the results discussed above.
On the 2D synchronous maps, peaks are symmetric with respect to the diagonal line in the correlation map. Peaks that appear along the diagonal are called “autopeaks”, which are always positive, indicating that the peak at the same wavenumber changes greatly under the external perturbation. The off-diagonal peaks (Φ(ν1, ν2)) are cross peaks, which may be positive or negative. The positive cross-peaks (Φ(ν1, ν2)) demonstrate that both peaks ν1 and ν2 change in the same direction (both increase or decrease) under the perturbation, whereas negative cross-peaks infer that the intensities of peaks ν1 and ν2 change in opposite directions (one increases while the other one decreases).
In 2D asynchronous spectra, there are no autopeaks but only off-diagonal cross-peaks, which can be either positive or negative. According to Noda's rule, if a cross-peak (ν1, ν2, and assume ν1 > ν2) has the same sign (both positive or negative) in the synchronous and asynchronous maps, the change of peak ν1 may occur prior to that of ν2, and vice versa. Hence, combined with synchronous and asynchronous spectra, some useful information about the temporal sequence of events can be obtained.
According to Noda's rule, the corresponding change order of PNIPAM in pure water during heating has been reported before,33 and is presented here for comparison. In addition, a sample of PNIPAM/D2O with 1.5 mol L−1 [Bmim][BF4] shows a slight variation in the temperature elevation, which suggests the existence of a stable structure. Therefore, we take the 0.4 mol L−1 IL sample as an example to analyze the thermally induced evolution of various microstructures at low IL concentration by the 2D correlation method, and to further investigate the role of ionic liquid in the phase separation of aqueous PNIPAM solutions.
A. The ν(C–H) region of 3020–2840 cm−1. For the PNIPAM C–H groups in pure water during heating, two-step side-chain dehydration first emerges, specifically, the CH3 groups hydrated with more water molecules change prior to the less hydrated ones, and then the main-chain aggregation follows.33 However, for the 0.4 mol L−1 IL sample, according to the interesting phenomenon reflected by the variable-temperature spectra and PCMW map, there are three regions that occur during the heating: 25–32, 33–37, and 38–45 °C. As the first region is related to the relatively stable stage, we study 2D maps in the latter two regions hereafter.
(1) 33–37 °C. In the synchronous map of Fig. 9a, autopeaks exist in three main bands centered at 2989, 2968, and 2925 cm−1, indicating that these bands change greatly with the temperature variation. As seen in the asynchronous spectrum (Fig. 9b), several cross peaks appear, which suggests the asynchronism of responses given by corresponding molecular vibrations toward the thermal perturbation. However, some of them are spurious peaks produced by baseline variations or affected by the shift of bands, which can be excluded by comparison with 1DIR. Thus, results are gained according to Noda's rule, from which we could obtain the final specific order of the three main bands as follows (→means prior to or earlier than): 2968 → 2925 → 2989 cm−1. It is has been known that the assignment of peaks at higher wavenumber in the C–H region is the vibration of methyl groups hydrated with more or fewer water molecules, however, in accordance with our previous confirmation, the addition of IL can destroy the hydration structure by preferentially interacting with D2O. Therefore, the peaks at 2868 cm−1 can be attributed to the vibration of the CH3 groups hydrated with fewer D2O molecules, while the peak at 2989 cm−1 could be assigned to the CH3 groups interacting with the IL–D2O association. Furthermore, the 2925 cm−1 peak originates from the CH2 groups on the backbone of PNIPAM.33 The discerned transform order reveals that during heating, the side-chain C–H groups dehydrate first, and then the main-chain aggregates, followed by the dissociation of the structure composed of CH3 groups and IL–D2O association. In this period, the phase transition takes place at a lower temperature than PNIPAM in pure water, mainly due to the one step, not the two step dehydration process during heating.33
![2D synchronous (a) and asynchronous (b) spectra of PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] in ν(C–H) region during heating generated from all the spectra at 33 to 37 °C. Warm colors (red) refer to positive intensities, while cold color (green) to negative ones.](/image/article/2012/RA/c2ra01349e/c2ra01349e-f9.gif) | ||
| Fig. 9 2D synchronous (a) and asynchronous (b) spectra of PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] in ν(C–H) region during heating generated from all the spectra at 33 to 37 °C. Warm colors (red) refer to positive intensities, while cold color (green) to negative ones. | ||
(2) 38–45 °C. As presented in 1DIR, interestingly, the intensities of the C–H groups gradually decrease in the range of 38–45 °C, which is above the LCST, so we can conclude that the changes should not be due to intra- or intermolecular hydrogen bonding between polymer segments, but may be related to the IL–D2O association. 2D maps, synchronous and asynchronous, in the ν(C–H) region of the PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] during heating from 38 to 45 °C are shown in Fig. 10. Similarly, the sequence can be extracted as follows: 2987 → 2965 → 2927 cm−1. With the 2987, 2965, and 2927 cm−1 bands assigned to vas(CH3) attached to the imidazole ring of [Bmim][BF4], vas(CH3) in the terminal CH3 groups of butyl side-chain, and vas(CH2) associated to the CH2 groups of butyl side-chain,43 the sequence may reflect the steps during dehydration of the IL alkyl group: the free CH3 groups change prior to the terminal CH3 groups, followed finally by the CH2 groups. Meanwhile, the aggregate size of PNIPAM solutions with IL is expected to be larger than that in IL-free solution,22 which is the evidence that IL–D2O association may take part in the formation of a globule, and as temperature elevates, the association may be damaged.
![2D synchronous (a) and asynchronous (b) spectra of PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] in ν(C–H) region during heating generated from all the spectra at 38 to 45 °C. Warm colors (red) refer to positive intensities, while cold color (green) to negative ones.](/image/article/2012/RA/c2ra01349e/c2ra01349e-f10.gif) | ||
| Fig. 10 2D synchronous (a) and asynchronous (b) spectra of PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] in ν(C–H) region during heating generated from all the spectra at 38 to 45 °C. Warm colors (red) refer to positive intensities, while cold color (green) to negative ones. | ||
B. The ν(C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) O) region of 1680–1588 cm−1.
As seen from Fig. 11, we can conclude the order of change is: 1656 → 1625 → 1610 → 1645cm−1, that is, v(CO⋯IL) → v(CO⋯D–O–D) → v (CO⋯D2O–IL) → v(CO⋯D–N) . Namely, the weaker CO⋯IL hydrogen bond (oxygen atoms of CO groups H–bonded with C–H groups of imidazole ring) introduced by the IL addition44,45 is broken prior to the hydrogen bond of CO⋯D–O–D. After that the strongest CO⋯D2O–IL hydrogen bond is destroyed as temperature increases, and finally, the carbonyl occurs in the form of CO⋯D–N. At lower IL concentration, because of the unstable CO⋯IL hydrogen bonds, the carbonyl will remove such a hydrogen bond earlier as a result of the thermal perturbation. Whereas, at the higher ratio, as previously confirmed, CO⋯D–N hydrogen bonds form at room temperature, together with CO⋯IL, CO⋯D–O–D, and CO⋯D2O–IL, leading to the stable network interaction via intra- and intermolecular hydrogen bonding, which is unaffected by the variation of temperature.
O) region of 1680–1588 cm−1.
As seen from Fig. 11, we can conclude the order of change is: 1656 → 1625 → 1610 → 1645cm−1, that is, v(CO⋯IL) → v(CO⋯D–O–D) → v (CO⋯D2O–IL) → v(CO⋯D–N) . Namely, the weaker CO⋯IL hydrogen bond (oxygen atoms of CO groups H–bonded with C–H groups of imidazole ring) introduced by the IL addition44,45 is broken prior to the hydrogen bond of CO⋯D–O–D. After that the strongest CO⋯D2O–IL hydrogen bond is destroyed as temperature increases, and finally, the carbonyl occurs in the form of CO⋯D–N. At lower IL concentration, because of the unstable CO⋯IL hydrogen bonds, the carbonyl will remove such a hydrogen bond earlier as a result of the thermal perturbation. Whereas, at the higher ratio, as previously confirmed, CO⋯D–N hydrogen bonds form at room temperature, together with CO⋯IL, CO⋯D–O–D, and CO⋯D2O–IL, leading to the stable network interaction via intra- and intermolecular hydrogen bonding, which is unaffected by the variation of temperature.
![2D synchronous (a) and asynchronous (b) spectra of PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] in ν(CO) region during heating generated from all the spectra at 25 to 45 °C. Warm colors (red) refer to positive intensities, while cold color (green) to negative ones.](/image/article/2012/RA/c2ra01349e/c2ra01349e-f11.gif) | ||
| Fig. 11 2D synchronous (a) and asynchronous (b) spectra of PNIPAM/D2O solution with 0.4 mol L−1 [Bmim][BF4] in ν(CO) region during heating generated from all the spectra at 25 to 45 °C. Warm colors (red) refer to positive intensities, while cold color (green) to negative ones. | ||
3.3 Proposed role of IL dynamics on phase behavior of aqueous PNIPAM solution
On the basis of the above analysis, we can clearly understand the influence of IL on the phase separation of aqueous PNIPAM solutions during heating. For a more intuitive understanding of the microdynamic mechanism, all proved and assigned structures are illustrated in Fig. 12. Interestingly, the addition of IL can modify the phase behavior of the polymer in water. At low IL concentrations, the LCST of aqueous PNIPAM solutions decreases with increasing IL content, while at higher IL concentrations, the phase transition disappears. The ionic liquid plays two opposite roles, the “destroyer” and the “constructer”, with variations in the concentration and temperature.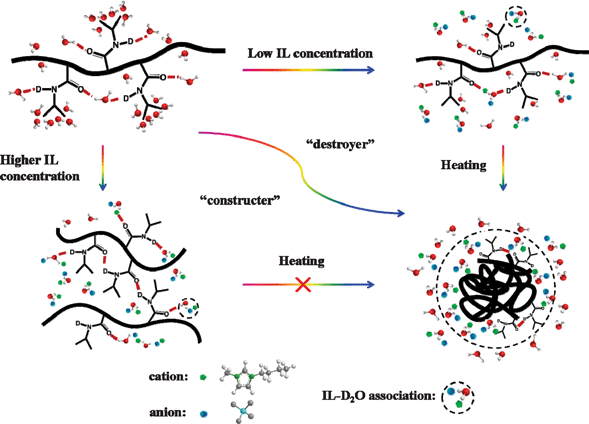 | ||
| Fig. 12 Schematic illustration of role of ionic liquid dynamics on the phase separation of aqueous PNIPAM solutions with variation in the IL concentration and temperature. | ||
At low IL concentrations in the aqueous PNIPAM solution, the IL prefers to interact with water molecules, leading to destabilization of the hydrated macromolecule structure via destruction of the hydration effect in the C–H and CO groups. As the temperature elevates, two changes occur: the former is related to the phase separation of PNIPAM solution, while the latter is attributed to the IL–D2O association, where the IL–D2O association takes part in the globule construction, probably interacting with hydrophilic groups of PNIPAM (CO groups), and assembling at the surface of collapsed globular structure. In this case, the role of IL is the “destroyer” for destabilization of the hydrated macromolecule structure below LCST. The IL acts as the “constructer” for globule formation above the LCST (in the form of IL–D2O association).
On the other hand, with more addition of IL, the damage of hydration effect and hydrogen bonds between the CO groups of the polymer and water molecules is further facilitated. Thus, the intra- and intermolecular hydrogen bonding CO⋯D–N form at room temperature, together with the CO⋯IL, CO⋯D–O–D, and CO⋯D2O–IL, leading to the formation of a stable interaction network among PNIPAM, water molecules, and ionic liquid, which is unaffected by the variation of temperature. Namely, the LCST phase behavior disappears. Thus, in this case, the ionic liquid could be regarded as the “constructer” during heating, which is a steady role in the interaction network.
Conclusions
In our work, FTIR in combination with two-dimensional correlation spectroscopy (2Dcos) and perturbation correlation moving window (PCMW) technique was employed for the first time to elucidate the role of [Bmim][BF4] dynamics on transition behavior of concentrated PNIPAM solutions (5% (w/v)). This was achieved through the analysis of two main regions (the C–H stretching band (3020–2840 cm−1), and the amide I (CO hydrogen bonding) (1680–1588 cm−1)).
It is interesting that the addition of IL can modify phase transition behavior of the aqueous PNIPAM solution: at low IL concentrations, the Tp of PNIPAM solutions decreases with an increase in the IL concentration, due to destabilization of the hydrated macromolecule structure; however, at higher IL concentrations, unexpectedly, the phase transition behavior disappears. This was attributed to the formation of a stable interaction network via intra- and intermolecular hydrogen bonding, such as the intermolecular hydrogen bonds of CO⋯D–N at low temperature, resulting from the hydrophobic collapse caused by the IL, not a response to the thermal perturbation.
As the temperature elevates, the sample with 0.4 mol L−1 IL shows two changes in the ν(C–H) region for the bands of vas(CH3) and vas(CH2). First a red shift and an intensity increase occur, which are related to the LCST phase behavior of PNIPAM solution. An obvious decrease in the intensities of the two peaks then occurs, which is relative to the IL–D2O association. In the former step, IL molecules damage the hydration effect of the C–H groups via H–bonded with water molecules preferentially; in the latter, the IL–D2O association takes part in the globule construction, probably via interaction with the hydrophilic groups of PNIPAM (CO groups). Thus, the role of IL dynamics on the phase behavior is mainly embodied in two opposite aspects, the “destroyer”, as well as the “constructer”.
Acknowledgements
We gratefully acknowledge financial support from the National Science Foundation of China (NSFC) (20934002) and the National Basic Research Program of China (No. 2009CB930000).References
- J. Kobayashi, A. Kikuchi, K. Sakai and T. Okano, Anal. Chem., 2003, 75, 3244 CrossRef CAS.
- S. J. Ahn, M. Kaholek, W. K. Lee, B. LaMattina, T. H. LaBean and S. Zauscher, Adv. Mater., 2004, 16, 2141 CrossRef CAS.
- S. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905 CrossRef CAS.
- Y. Ding, X. Ye and G. Zhang, Macromolecules, 2005, 38, 904 CrossRef CAS.
- K. Zhou, Y. Lu, J. Li, L. Shen, G. Zhang, Z. Xie and C. Wu, Macromolecules, 2008, 41, 8927 CrossRef CAS.
- N. Al-Manasir, K. Zhu, A. L. Kjøniksen, K. D. Knudsen, G. Karlsson and B. Nyström, J. Phys. Chem. B, 2009, 113, 11115 CrossRef CAS.
- F. Meersman, J. Wang, Y. Wu and K. Heremans, Macromolecules, 2005, 38, 8923 CrossRef CAS.
- S. C. Jung, S. Y. Oh and Y. Chan Bae, Polymer, 2009, 50, 3370 CrossRef CAS.
- H. Yamauchi and Y. Maeda, J. Phys. Chem. B, 2007, 111, 12964 CrossRef CAS.
- G. Dalkas, K. Pagonis and G. Bokias, Polymer, 2006, 47, 243 CrossRef CAS.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, W. Pan, M. Garcia-Garibay and N. J. Turro, Macromolecules, 1993, 26, 4577 CrossRef CAS.
- F. M. Winnik, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415 CrossRef CAS.
- L. T. Lee and B. Cabane, Macromolecules, 1997, 30, 6559 CrossRef CAS.
- B. Jean, L. T. Lee and B. Cabane, Langmuir, 1999, 15, 7585 CrossRef CAS.
- W. Loh, L. A. C. Teixeira and L. T. Lee, J. Phys. Chem. B, 2004, 108, 3196 CrossRef CAS.
- B. Jean and L. T. Lee, J. Phys. Chem. B, 2005, 109, 5162 CrossRef CAS.
- V. Jijo, K. P. Sharma, R. Mathew, S. Kamble, P. Rajamohanan, T. Ajithkumar, M. Badiger and G. Kumaraswamy, Macromolecules, 2010, 43, 4782 CrossRef CAS.
- A. C. Kumar, H. Erothu, H. B. Bohidar and A. K. Mishra, J. Phys. Chem. B, 2010, 115, 433 CrossRef.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505 CrossRef CAS.
- R. Freitag and F. Garret-Flaudy, Langmuir, 2002, 18, 3434 CrossRef CAS.
- K. Suwa, K. Yamamoto, M. Akashi, K. Takano, N. Tanaka and S. Kunugi, Colloid Polym. Sci., 1998, 276, 529 CAS.
- P. M. Reddy and P. Venkatesu, J. Phys. Chem. B, 2011, 115, 4752 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431 CrossRef CAS.
- P. Kubisa, Prog. Polym. Sci., 2009, 34, 1333 CrossRef CAS.
- T. Ueki and M. Watanabe, Chem. Lett., 2006, 35, 964 CrossRef CAS.
- T. Ueki and M. Watanabe, Langmuir, 2007, 23, 988 CrossRef CAS.
- I. Noda, J. Am. Chem. Soc., 1989, 111, 8116 CrossRef CAS.
- I. Noda, J. Mol. Struct., 2010, 974, 3 CrossRef CAS.
- M. Thomas and H. H. Richardson, Vib. Spectrosc., 2000, 24, 137 CrossRef CAS.
- S. Morita, H. Shinzawa, I. Noda and Y. Ozaki, Appl. Spectrosc., 2006, 60, 398 CrossRef CAS.
- B. Sun, Y. Lin, P. Wu and H. W. Siesler, Macromolecules, 2008, 41, 1512 CrossRef CAS.
- S. Sun, J. Hu, H. Tang and P. Wu, J. Phys. Chem. B, 2010, 114, 9761 CrossRef CAS.
- B. Sun, Y. Lin and P. Wu, Appl. Spectrosc., 2007, 61, 765 CrossRef CAS.
- S. Sun and P. Wu, Macromolecules, 2010, 43, 9501 CrossRef CAS.
- P. Schmidt, J. Dybal and M. Trchová, Vib. Spectrosc., 2006, 42, 278 CrossRef CAS.
- M. López-Pastor, María José Ayora-Cañada, M. Valcárcel and B. Lendl, J. Phys. Chem. B, 2006, 110, 10896 CrossRef.
- B. Sun, Q. Jin, L. Tan, P. Wu and F. Yan, J. Phys. Chem. B, 2008, 112, 14251 CrossRef CAS.
- L. Zhang, Z. Xu, Y. Wang and H. Li, J. Phys. Chem. B, 2008, 112, 6411 CrossRef CAS.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325 CrossRef CAS.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503 CrossRef CAS.
- J. Kiefer, J. Fries and A. Leipertz, Appl. Spectrosc., 2007, 61, 1306 CrossRef CAS.
- C. Jiang, S. C. Li, P. M. Shih, T. C. Hung, S. C. Chang, S. H. Lin and H. C. Chang, J. Phys. Chem. B, 2010, 115, 883 CrossRef.
- Z. Wang and P. Wu, J. Phys. Chem. B, 2011, 115, 10604 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra01349e/ |
| This journal is © The Royal Society of Chemistry 2012 |