Photocatalytic reduction of CO2 with H2O on various titanium oxide photocatalysts
Kohsuke
Mori
a,
Hiromi
Yamashita
*a and
Masakazu
Anpo
*b
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.jp; Fax: +81-6-6879-7457
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka Prefecture University, 1-1 Gakuen-cho, Naka-ku, Sakai, Osaka 599-8531, Japan. E-mail: anpo@chem.osakafu-u.ac.jp; Fax: +81-72-254-9941; Tel: +81-72-254-9139
First published on 11th January 2012
Abstract
The specific features of the photocatalytic reduction of CO2 with H2O on various types of active titanium oxide catalysts are reviewed. UV-light irradiation of the bulk TiO2 powders in the presence of CO2 and H2O at room temperature under heterogeneous gas-solid conditions produced CH4 as the major product, while the predominant formations of CH3OH as well as CH4 were observed on the highly dispersed titanium oxide moiety anchored on zeolites and mesoporous silica materials. The CH3OH formation is originated from the unique properties of the charge transfer excited state, i.e., (Ti3+–O−)* of the tetrahedrally-coordinated titanium oxide species within the silica frameworks.
1. Introduction
Highly efficient and selective photocatalytic systems driven under sunlight and accompanied with a large positive change in the Gibbs free energy are of vital interest.1–8 These reactions are considered to be an up-hill reaction and are similar to photosynthesis by green plants which produces glucose and oxygen from CO2 and H2O.The chemical conversion of CO2 into industrially beneficial compounds is also advantageous in terms of Green and Sustainable Chemistry because CO2 is an inexpensive, nontoxic and abundantC1 feedstock. Therefore, the development of efficient photocatalytic systems enabling the reduction and/or fixation of CO2 is a desirable and challenging goal.2,3The utilization of solar energy for the reduction and/or fixation of CO2 can be realized by considering the photocatalytic reduction and/or fixation of CO2 with H2O into CO, HCOOH, CH3OH, and CH4, etc. by active photocatalysts such as TiO2, SrTiO3, or SiC semiconductors. Inoue and Fujishima, et al.9 first reported that HCOOH, HCHO, and CH3OH are produced under irradiation of aqueous suspension systems involving a variety of semiconductor powders or single crystals. Although the pioneering works on the photo-reduction of CO2 on semiconductors in aqueous suspension systems were summarized by Halmann, the efficiency was low when H2O was used as the reductant.10 Substantially improved yields of CH3OH and CH4 from CO2 with H2O have been reported by us, in which photocatalytic reactions successfully proceed in solid-gas systems at room temperature on isolated tetrahedral Ti centers of macro- or mesoporous silicate sieves instead of dense phase powdered TiO2 materials.2,3,11–22
With these objectives of research in mind, we focus on the characteristic features of photocatalytic reduction and/or fixation of CO2 with H2O on various types of active titanium oxide catalysts under heterogeneous gas-solid conditions. Studies have been carried out on extremely small TiO2 particles and on highly dispersed anchored titanium oxide catalysts under UV-irradiation.11–22 Special attention has been paid to the relationship between the local structure of the active Ti site and their photocatalytic performances. Additionally, investigations on the dynamic properties of the excited state of active Ti species as well as on the direct detection of the reaction intermediate species have been summarized in order to gain an insight into the molecular level reaction mechanism.
2. Photocatalytic reactions on various titanium oxide catalysts
As shown in Fig. 1, the electronic properties as well as the photocatalytic activity of titanium oxide photocatalysts dramatically changes depending on their local structure, from an extended-semiconducting structure to nano-sized, extremely small nanoparticles and molecular-sized titanium oxide species. In the case of the bulk TiO2 materials, photochemical excitation leads to charge separation in the particle, in which electrons (e−) are promoted to the conduction band (cb) and holes (h+) are left in the valence band (vb) upon light irradiation. The generated hole in the valence band and the conduction band electron can react with electron donors and electron acceptors adsorbed on the titanium oxide surface, respectively. On the contrary, the Ti oxide moieties, which are spatially separated each other, can be implanted and isolated in the silica matrixes of microporous zeolite and mesoporous silica materials at the atomic level and have been called “single-site photocatalysts”. The isolated Ti atoms can be substituted with Si atoms in the silica matrixes (Si/Ti > 30) and coordinated tetrahedrally with oxygen atoms. Such single-site catalysts can be simply synthesized by various anchoring techniques such as hydrothermal synthesis, sol–gel method and chemical vapor deposition, etc., and are stable as long as the silica matrixes keep their porous structures. In the case of an isolated and tetrahedrally coordinated Ti–oxide moiety, excitation by UV irradiation brings about an electron transfer from the oxygen (O2−) to Ti4+ ions, resulting in the formation of pairs of trapped hole centers (O−) and electron centers (Ti3+). Such a charge transfer excited state, i.e., the excited electron-hole pair state which localize quite near to each other compared to the electron and hole produced in semiconducting materials, plays a significant role in various photocatalytic reactions.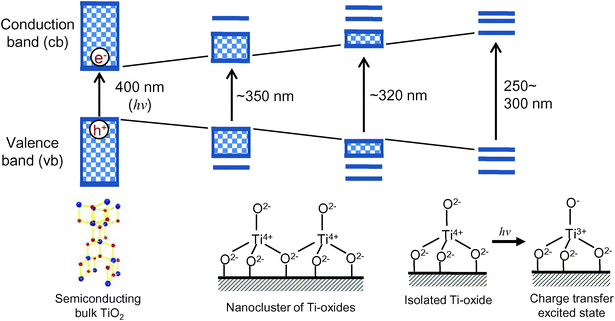 | ||
| Fig. 1 The electronic state change in the titanium oxide photocatalysts from semiconducting bulk TiO2 to isolated Ti–Oxide molecular species. | ||
The efficiency for the photocatalytic reduction and/or fixation of CO2 with H2O to produce CH4 and CH3OH strongly depends upon the type of the employed titanium oxide photocatalysts.11–22 The primary processes on semiconducting TiO2 photocatalysts is illustrated in Fig. 2. The band gap between the conduction band and the valence band becomes larger as the particle size of the semiconducting TiO2 decreases, making it suitable and applicable for the reduction of CO2.2,3 For extremely fine TiO2 particles less than 10 nm in diameter, the size quantum effect and/or the effects of the surface modification in its coordination geometry plays a significant role in the appearance of unique activity. Consequently, the electrons and holes which are produced by UV-light irradiation within the ultrafine particles of TiO2 and the highly dispersed titanium oxide species exhibit more unique and high activities compared to those produced in large particle TiO2 photocatalysts.
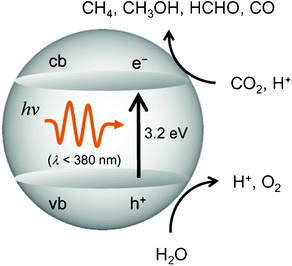 | ||
| Fig. 2 Reaction scheme for the photocatalytic reduction of CO2 with H2O on bulk TiO2. | ||
3. Small particle TiO2 catalysts
UV-irradiation of powdered TiO2 catalysts in the presence of a gaseous mixture of CO2 and H2O led to the evolution of CH4 at 275 K, accompanied by the formation of trace amounts of C2H4 and C2H6.13,14 The yields of these products increased with the UV-irradiation time, while no products were detected under dark conditions. The CH4 yield was almost zero in the reaction of CO2 without H2O, and increased when increasing the amount of H2O. These results suggest that the photocatalytic reduction of CO2 to produce CH4 and C2-compounds takes place photocatalytically in the solid–gas phase systems. The formation of CH4 as the main product has also been observed by Saladin et al., and the partially reduced TiO2 species formed under UV-irradiation is proposed as an active species.23The yields of CH4 formation in the photocatalytic reduction of CO2 with H2O on several TiO2 photocatalysts with different physicochemical property are shown in Table 1. The photocatalytic activity, based on the CH4 yields, was found to depend on the type of TiO2 catalyst, in the order of JRC-TIO-4 > -5 > -2 > -3.14 This tendency corresponds with that of the photocatalytic hydrogenation of methyl acetylene with H2O, proving that the reduction of CO2 with H2O undoubtedly occurs via a photocatalytically activated powdered TiO2 catalyst.24,25 It is likely that the anatase-type TiO2 possessing a large band gap as well as numerous surface –OH groups is preferable for efficient photocatalytic reactions. The band gap increase is accompanied by a shift in the conduction band edge to higher energy levels. This shift causes the reductive potential to shift to more negative values, which ultimately causes a great enhancement in the photocatalytic activity. The surface –OH groups and/or physisorbed H2O also play a significant role in photocatalytic reactions via the formation of OH radicals and H radicals.
| Catalyst (JRC-TIO-) | SBET (m2 g−1) | CO2 ads (μmol g−1) | Relative-OH conc | Band gap (eV) | Reduction of CO2a (μmol h−1 g) | Hydrogenation of methyl acetylene (μmol h−1 g) |
|---|---|---|---|---|---|---|
| a CH4 yield in the reaction of CO2 (0.12 mmol) and H2O (0.37 mmol) for 6 h. | ||||||
| 2 (anatase) | 16 | 1 | 1 | 3.47 | 0.03 | 0.20 |
| 3 (rutile) | 51 | 17 | 1.6 | 3.32 | 0.02 | 0.12 |
| 4 (anatase) | 49 | 10 | 3.0 | 3.50 | 0.27 | 8.33 |
| 5 (rutile) | 3 | 0.4 | 3.1 | 3.09 | 0.04 | 0.45 |
Preliminary studies by ESR measurement provide an insight into the reaction pathway. The ESR signals obtained under UV-irradiation of the anatase-type TiO2 catalyst in the presence of CO2 and H2O at 77 K can be ascribed to the characteristic photogenerated Ti3+ ions (g⊥ = 1.9723 and g║ = 1.9628) and H radicals (with 490 G splitting), as well as CH3 radicals having a hyperfine splitting (Hα = 19.2 G, g = 2.002).14 The signal intensity of CH3 radicals decreased when increasing the amount of H2O, indicating that CH3 radicals are the intermediate species and react with H radicals that are formed by the reduction of protons (H+) originated from H2O adsorbed on the catalyst.
4. Metal-loaded TiO2 powder
The effect of metal-loading on the photocatalytic reduction of CO2 with H2O on TiO2 catalysts was investigated.14 In the case of the Cu/TiO2 photocatalyst(0.3∼1.0 wt%), the CH4 yield was suppressed, but a new formation of CH3OH could be observed. Characterization by XPS reveals that the main species of copper in the catalyst is in the 1+ oxidation state. It has been also reported that Cu+ catalysts play a significant role in the photoelectrochemical production of CH3OH from CO2 and H2O system.26 Following the photocatalytic reduction, the Cu/TiO2 catalysts exhibited a new peak at 299 eV in the C(1) XPS spectra, suggesting that carboxylate groups which accumulated on the catalyst may be the primary intermediate species for this reaction.A further detailed study was performed by Gunlazuardi et al., by systematically varying the Cu loading on TiO2.27 The CH3OH yield increased with Cu loading and the highest yield can be achieved by 3% Cu/TiO2, which was 3 times higher than the original TiO2. Cu can serve as an electron trapper and prohibits the recombination of electron and hole, significantly increasing photoefficiency. However, catalysts with more than 3 wt% Cu loading do not further increase the CH3OH yield due to its shading effects, consequently reducing the photo-exciting capacity of TiO2. The activation energy for 3% Cu/TiO2 and the original TiO2 was determined to be 12 and 26 kJ mol−1, respectively. The apparent lower activation energy of 3% Cu/TiO2 catalyst indicates that the Cu also acts as an active species to provide CH3OH, and enhance the photoefficiency of TiO2photocatalysts.
In the case of thePt/TiO2,the yield of CH4 increased remarkably when the amount of Pt was increased (0.1 ∼ 1.0 wt%), but the addition of excess Pt was undesirable for an efficient reaction.13 Regarding the reaction intermediates, Solymosi et al. have observed the formation of CO2− species in bent form under UV-irradiation of the Rh/TiO2 in the presence of CO2 using FT-IR.28 The electron transfer from the irradiated catalyst to the adsorbed CO2 takes place, resulting in the formation of a CO2− anion as the key step in the photochemical reduction of CO2 using metal loaded TiO2 semiconductor.
5. TiO2 single crystals
With a well-defined catalyst surface such as a single crystal, detailed information on the reaction mechanism can be obtained at the molecular level.2,15 Therefore, the photocatalytic reduction of CO2 with H2O on rutile-type TiO2 (100) and TiO2(110) single crystal surfaces have been performed.15 As shown in Table 2, the efficiency and selectivity of the photocatalytic reactions strongly depend on the type of TiO2 single crystal surface. UV-irradiation of the TiO2(100) single crystal catalyst in the presence of a mixture of CO2 and H2O led to the evolution of both CH4 and CH3OH at 275 K, whereas only CH3OH was detected with the TiO2(110) single crystal catalyst.| Single crystal | Yield of CH4 (μmol h−1 g-cat) | Yield of CH3OH (μmol h−1 g-cat) |
|---|---|---|
| TiO2 (100) | 3.5 | 2.4 |
| TiO2 (110) | 0 | 0.8 |
It is likely that the photo-generated electrons localize on the surface sites of excited TiO2 play a significant role in the photoreduction of CO2 molecules into intermediate carbon species.2,15 The surface Ti atoms may act as a reductive site. According to the surface geometric models for TiO2(100) and TiO2(110), the atomic ratio (Ti/O) of the top-surface Ti and O atoms which have geometric spaces large enough to have direct contact with CO2 and H2O molecules, is higher on TiO2(100) than on TiO2 (110) surface. In the excited state, the surface with a higher Ti/O surface ratio, i.e. TiO2(100), exhibits a more reductive tendency than TiO2(110). Such a reductive surface allows a more facile reduction of CO2 molecules especially for the formation of CH4. In the HREELS spectrum of a clean TiO2(100) single crystal surface after UV irradiation in the presence of CO2 and H2O, two peaks due to the C–H stretching vibration of the CHx species and the O–H stretching of the surface hydroxyl groups at around 2920 and 3630 cm−1, respectively. On the other hand, only a weak peak assigned to the O–H stretching vibration was observed without UV irradiation, suggesting that UV-light irradiation is indispensable for attaining CO2 reduction and the formation of the active H and CHx species.
6. Ti–oxide anchored on zeolite (ion-exchange)
The photocatalyst systems incorporated within the zeolite cavities and frameworks have been proven to be effective for various reactions.29–32 The Ti–oxide anchored onto zeolite, Ti–oxide/Y-zeolite (1.1 wt% as TiO2), was prepared by ion-exchange with an aqueous titanium ammonium oxalate solution using Y-zeolite (SiO2/Al2O3 = 5.5) (ex-Ti–oxide/Y-zeolite).16–18Fig. 3 shows the Ti K-edge XANES and the Fourier transformation of EXAFS (FT-EXAFS) spectra of the Ti–oxide/Y-zeolites. The XANES spectra of the bulk TiO2 powder gives rise to several well-defined pre-edge peaks attributable to titanium in a symmetric octahedral environment.33 The ex-Ti–oxide/Y-zeolite exhibits an intense single pre-edge peak at 4967 eV suggesting that the Ti–oxide species exist in a tetrahedral coordination.1–3 On the other hand, the imp-Ti–oxide/Y-zeolite prepared by the impregnation exhibits three characteristic weak pre-edge peaks attributed to crystalline TiO2. The FT-EXAFS spectra of the ex-Ti–oxide/Y-zeolite exhibits only peak at around 1.6 Å assigned to the neighboring oxygen atoms (Ti–O) indicating the presence of an isolated Ti–oxide species. These findings indicate that highly dispersed isolated tetrahedral Ti–oxide species are formed on the ex-Ti–oxide/Y-zeolite. On the other hand, the imp-Ti–oxide/Y-zeolite exhibits an intense peak assigned to the neighboring titanium atoms (Ti–O–Ti) suggestive of the aggregation of the Ti–oxide species.
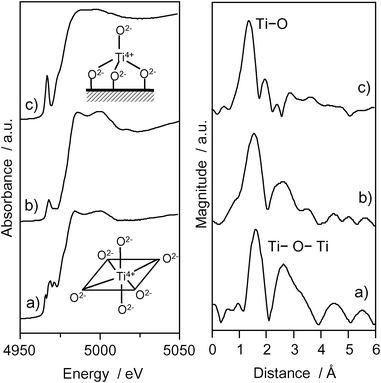 | ||
| Fig. 3 Ti K-edge XANES and FT-EXAFS spectra of anatase TiO2 powder (a), imp-Ti–oxide/Y-zeolite (10.0 wt% as TiO2) (b), and the ex-Ti–oxide/Y-zeolite (c). | ||
The photoluminescence spectra of the ex-Ti–oxide/Y-zeolite at 77 K are shown in Fig. 4. Excitation by light at around 250–280 nm brought about an electron transfer from the oxygen to titanium ion, resulting in the formation of pairs of the trapped hole center (O−) and an electron center (Ti3+).1–3 The observed photoluminescence is attributed to the radiative decay process from the charge transfer excited state of the Ti–oxide moieties in a tetrahedral coordination geometry, (Ti3+–O−)*, to their ground state.34,35 The addition of H2O or CO2 molecules onto the anchored Ti–oxide species leads to the efficient quenching of the photoluminescence. Such an efficient quenching suggests not only that tetrahedrally coordinated Ti–oxide species locate at positions accessible to the added CO2 or H2O but also that CO2 or H2O interacts and/or reacts with the Ti–oxide species in both its ground and excited states. Because the addition of CO2 led to a less effective quenching than with the addition of H2O, the interaction of the emitting sites with CO2 was weaker than with H2O.
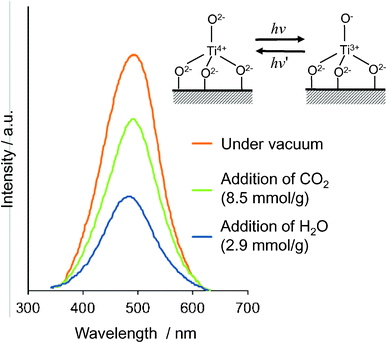 | ||
| Fig. 4 Photoluminescence spectrum of the ex-Ti–oxide/Y-zeolite catalyst (a), and the effects of the addition of CO2 and H2O. Measured at 77 K, excitation at 290 nm, emission monitored at 490 nm. | ||
UV-irradiation of powdered TiO2 and Ti–oxide/Y-zeolite catalysts in the presence of a mixture of CO2 and H2O led to the evolution of CH4 and CH3OH at 328 K, as well as trace amounts of CO, C2H4 and C2H6.16–18 The specific photocatalytic activities for the formation of CH4 and CH3OH are shown in Fig. 5. The ex-Ti–oxide/Y-zeolite exhibits a high activity and a high selectivity for the formation of CH3OH, while the formation of CH4 was predominated on bulk TiO2 as well as on the imp-Ti–oxide/Y-zeolite. The deposition of Pt improved the photocatalytic activity, but the CH3OH selectivity significantly decreased. These findings clearly suggest that the tetrahedrally coordinated Ti–oxide species act as active photocatalysts for the reduction of CO2 with H2O exhibiting a high selectivity toward CH3OH.
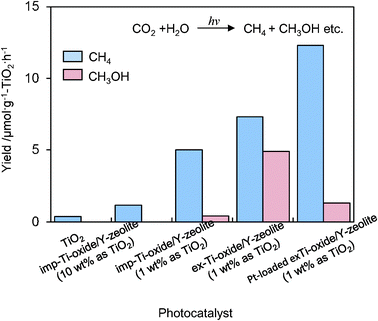 | ||
| Fig. 5 The product distribution of the photocatalytic reduction of CO2 with H2O on anatase TiO2 powder, imp-Ti–oxide/Y-zeolite (10 wt% as TiO2), imp-Ti–oxide/Y-zeolite (1 wt% as TiO2), the ex-Ti–oxide/Y-zeolite (1 wt% as TiO2), and the Pt-loaded ex-Ti–oxide/Y-zeolite catalysts. | ||
UV-irradiation of the anchored Ti–oxide catalyst in the presence of CO2 and H2O at 77 K led to the appearance of ESR signals due to the Ti3+ions, H atoms, and carbon radicals.2,3 After the disappearance of these ESR signals in this system at around 275 K, the formation of CH4 and CH3OHwas observed. From these results, the reaction mechanism in the photocatalytic reduction of CO2 with H2O on the highly dispersed Ti–oxide catalyst can be proposed as shown in Scheme 1: CO2 and H2O molecules interact with the excited state of the photoinduced (Ti3+–O−)* species and the reduction of CO2 and the decomposition of H2O proceed competitively. Furthermore, H atoms and OH• radicals are formed from H2O and such radicals react with the carbon species formed from CO2 to produce CH4 and CH3OH.
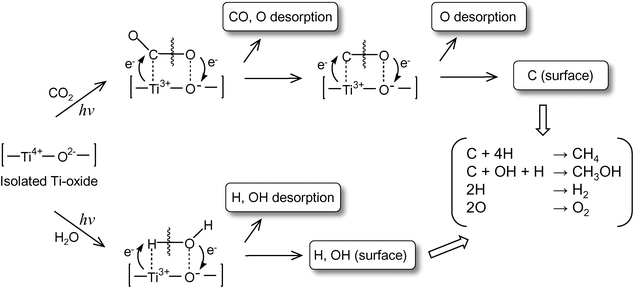 | ||
| Scheme 1 Schematic illustration of the photocatalytic reduction of CO2 with H2O on the anchored titanium oxide species. | ||
7. Ti-containing zeolite and mesoporous molecular sieves
The Ti–oxide species prepared within the ordered silica frameworks have revealed a unique local structure as well as a high selectivity in the oxidation of organic substances with hydrogen peroxide.34–36 Hydrothermally synthesized Ti-containing zeolites (TS-1, Ti–Beta) and mesoporous molecular sieves (Ti–MCM, Ti–HMS, Ti–FSM) have been subjected to the photocatalytic reduction CO2 with H2O.18–22,36–38In situ photoluminescence, ESR, UV-VIS and XAFS investigations indicated that the Ti–oxide species in the Ti-mesoporous molecular sieves (Ti–MCM-41 and Ti–MCM-48) and in the TS-1 zeolite are highly dispersed within the zeolite framework and exist in a tetrahedral coordination. Upon excitation with UV light at around 250–280 nm, these catalysts exhibit photoluminescence spectra at around 480 nm. The addition of CO2 or H2O onto these catalysts results in a significant quenching of the photoluminescence, suggesting the excellent accessibility of the CO2 and H2O to the Ti–oxide species.18–21
UV-irradiation of the Ti-mesoporous molecular sieves and the TS-1 zeolite in the presence of CO2 and H2O also led to the formation of CH3OH and CH4 as the main products.18–21 The yields of CH3OH and CH4 per unit weight of the Ti-based catalysts are shown in Fig. 6. It can be seen that Ti–MCM-48 exhibits much higher activity than either TS-1 or Ti–MCM-41. The higher activity and selectivity for the formation of CH3OH attained with the Ti–MCM-48 than with the other catalysts may be due to the combined contribution of the high dispersion state of the Ti–oxide species and the large pore size with a three-dimensional channel structure: TS-1 has a smaller pore size (ca. 5.7 Å) and a three-dimensional channel structure; Ti–MCM-41 has a large pore size (>20 Å) but one-dimensional channel structure; and Ti–MCM-48 has both a large pore size (>20 Å) and three-dimensional channels. These results strongly suggest that mesoporous molecular sieves containing highly dispersed Ti–oxide species are promising candidates as effective photocatalysts.
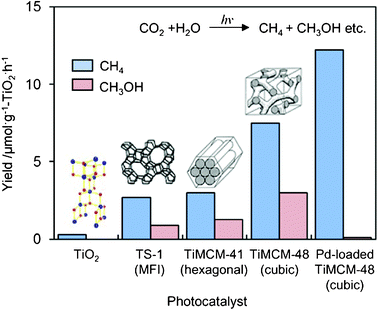 | ||
| Fig. 6 The product distribution of the photocatalytic reduction of CO2 with H2O on TiO2 powder, TS-1, Ti–MCM-41, Ti–MCM-48, and the Pt-loaded Ti–MCM-48 catalysts. | ||
The effect of Pt-loading on the photocatalytic activity of Ti-containing mesoporous silica has also been investigated and the changes in the yields of CH4 and CH3OH formation are shown in Fig. 6. The deposition of Pt onto Ti-containing zeolites is effective for promoting the photocatalytic activity, but only the formation of CH4 is promoted.18
The Ti-containing zeolite, Ti–Beta, has attracted much attention because of its large-pore structure compared to the Y-zeolite.36–38 The other characteristic feature is the H2O affinity of Ti–Beta zeolites can be changed significantly depending on the preparation methods, and their hydrophobic-hydrophilic properties can modify the catalytic performances.36–38 As shown in Fig. 7, the photocatalytic reduction of CO2 with H2O was found to proceed in the gas phase at 323 K with different activity and selectivity on hydrophilic Ti–Beta(OH) and hydrophobic Ti–Beta(F) zeolites prepared in OH− and F− media, respectively. The higher activity for the formation of CH4 observed with Ti–Beta(OH) and the higher selectivity for the formation of CH3OH observed with the Ti–Beta(F) may be attributed to the different abilities of zeolite pores on the H2O affinity. These results suggest that the affinity of the H2O molecules to adsorb on the zeolite is an important factor in the selectivity of the photocatalytic reduction of CO2 and H2O.
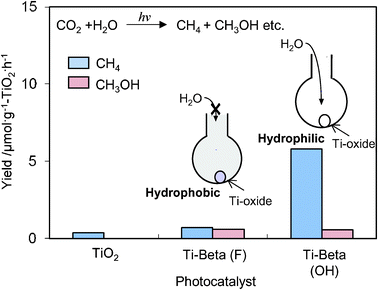 | ||
| Fig. 7 The product distribution of the photocatalytic reduction of CO2 with H2O on Ti–Beta(F), Ti–Beta(OH), and TiO2 powder (P-25) as the reference catalyst. | ||
For the reduction of CO2 with H2O on the Ti-containing zeolite and mesoporous molecular sieves, a similar reaction pathway with Ti–oxide anchored on zeolite is proposed by considering ESR spectroscopy at 77 K, in which CO2 reduction and H2O splitting proceed competitively at the LMCT-excited Ti–O centers: CO2 is reduced to CO, and subsequently to C radicals while H2O photodecomposes to H and OH radicals. The reaction of H and OH radicals with carbon species is thought to yield CH3OH and CH4. Frei and co-workers carried out further mechanistic investigations by monitoring the visible light-induced reaction of 13CO2 and H2O gas mixtures in a framework Ti–MCM-41 molecular sieve with in situ FT-IR spectroscopy at room temperature.3912CO gas along with 13CO were observed as the products by infrared, and the growth of 13CO depended linearly on the photolysis laser power. This points out the presence of small amounts of carbonaceous residues on the high-surface area mesoporous silicates. H2O was also confirmed as the stoichiometric electron donor. These results suggest that CO is a single-photon, 2-electron-transfer product of CO2 at framework Ti centers with H2O acting as a direct electron donor. By considering this, the mechanism is proposed as follows: Excitation of the framework Ti4+ centers leads to photoinduced (Ti3+—O−)* species. Electron transfer from transient Ti3+ to CO2 splits the molecule into CO and O−.The latter is spontaneously protonated by a Si–OH group, or H+ co-generated upon H2O oxidation to yield a surface OH radical. Another surface OH radical is formed as a result of the simultaneous H2O oxidation by the framework oxygen hole. The OH radicals either combine to yield H2O2 or dismutate to give O2 and H2O.
8. Ti–oxide anchored on porous silica glass (CVD)
The Ti–oxide anchored onto porous silica glass (PVG) plate was prepared using a facile reaction of TiCl4 with the surface OH groups on the transparent porous Vycor glass (Corning code 7930) in the gas phase at 453-473 K, followed by treatment with H2O vapor to hydrolyze the anchored compound.11–13 UV-irradiation of the anchored Ti–oxide catalysts in the presence of a mixture of CO2 and H2O led to the evolution of CH4, CH3OH and CO at 323 K. The total yield was larger under UV-irradiation at 323 K than at 275 K. The efficiency of the photocatalytic reaction strongly depends on the ratio of H2O/CO2 and its activity increases when increasing the H2O/CO2 ratio; however, an excess amount of H2O suppresses the reaction rates. Fig. 8 shows the effect of the number of anchored Ti–O layers on the absorption edge in the UV-vis spectra of the catalysts and the efficiency of the photocatalytic reactions as well as the relative yields of the photoluminescence. It was proven that only catalysts with highly dispersed monolayer Ti–oxide exhibit high photocatalytic activity and photoluminescence at around 480 nm. Only the tetrahedrally coordinated Ti–oxide species exhibit photoluminescence emission upon excitation at around 250–280 nm. These findings also clearly suggest that the tetrahedrally coordinated Ti–oxide species function as active photocatalysts for the reduction of CO2 with H2O.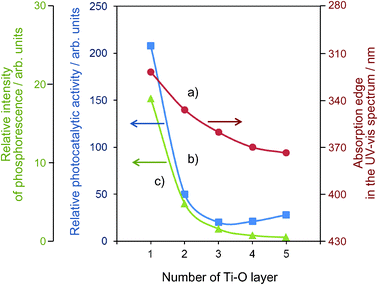 | ||
| Fig. 8 The effects of the number of the Ti–O layers of the anchored titanium oxide catalysts on the absorption edge of the catalysts (a), the reaction yields (b), and the relative yields of the photoluminescence (c). | ||
9. Ti-containing porous silica thin film
In the syntheses of zeolites and mesoporous molecular sieves, the morphology of these materials was mainly in powdered form. However, powdered material systems are difficult to handle for practical use as photocatalysts. Therefore, the synthesis of transparent porous silica thin films is a subject of current interest. Such transparent porous silica thin films have a larger surface area, in contrast to metal oxide thin films on a quartz substrate, and can realize the efficient absorption of light, showing that they have potential for use as effective photocatalysts.Self-standing Ti-containing porous silica (PS) thin films with different pore structures and Ti contents were synthesized by a solvent evaporation method. Ti–PS(h, 25) and Ti–PS(c, 50) mean hexagonal and cubic structure with a Si/Ti ratio of 25 and 50, respectively.40,41 The synthesized thin films are colorless and completely transparent. A mesoporous structure was confirmed by a characteristic diffraction peak at around 2–3° associated with the d100 spacing.
UV irradiation of these Ti-containing porous silica thin films in the presence of CO2 and H2O at 323 K led to the formation of CH4 and CH3OH as well as CO and O2 as minor products. Ti–PS(h, 50) thin film was proven to be a most effective photocatalyst with a high quantum yield of 0.28%, which was larger than the Ti–MCM-41 powder catalyst even with the same pore structure (Fig. 9). The quantum yield obtained with the transparent Ti–PS (50) thin film photocatalyst is improved in comparison to the value obtained for titanium oxide anchored on transparent porous silica glass (0.02%). In powdered form, the effect of the scattering of light on the particle surface would be large so that effective light absorption and measurement may not be realized. Thus the high photocatalytic activity of such thin films can be attributed to the efficient absorption of UV light due to its high transparency. From FT-IR investigations, it was found that these Ti–containing porous silica thin films had different concentrations of surface OH groups and showed different adsorption properties for the H2O molecules toward the catalyst surface: photocatalysts with small amounts of surface OH groups showed high selectivity for the formation of CH3OH.
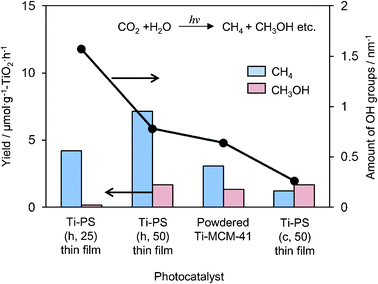 | ||
| Fig. 9 The product distribution of the photocatalytic reduction of CO2 with H2O and amount of surface OH groups on Ti–PS(h, 25), Ti–PS(h, 50), powdered Ti–MCM-41, and Ti–PS(c, 50). | ||
10. Ti/Si binary oxide (sol–gel)
Ti/Si binary oxides involving different Ti contents were prepared by the sol–gel method using mixtures of tetraethylorthosilicate and titanium-iso-propoxide. Ti/Si binary oxides with only a small Ti contents exhibited the photoluminescence emission at around 480 nm upon excitation at around 280 nm.22,42 UV-irradiation of the Ti/Si binary oxide catalysts in the presence of a gaseous mixture of CO2 and H2O led to the formation of CH4 and CH3OH as the main products. A parallel relationship between the specific photocatalytic activities of the titanium oxide species and the photoluminescence yields of the Ti/Si binary oxides was observed. This phenomenon clearly indicates that the appearance of high photocatalytic activity for the binary oxides is closely associated with the formation of the charge transfer excited complex due to the highly dispersed tetrahedral Ti–oxide species.The XAFS, ESR and photoluminescence investigations of the Ti/Si binary oxide indicated that these catalysts prepared by the sol–gel method can keep the tetrahedral coordination geometry of the Ti–oxide species until the TiO2 content approaches up to approximately 20 wt%. Consequently, the Ti/Si binary oxides with a high Ti content can be successfully utilized as active photocatalysts for the efficient reduction of CO2 with H2O in the gas-solid system.
11. Visible-light sensitive TiO2 based catalysts
The development of visible-light sensitive photocatalysts was intensively pursued since the pure TiO2 and Ti–oxide species only show fascinating photocatalytic activities under UV-light irradiation, which limits the utilization of a small UV fraction of natural solar light. One of the most promising strategies is to use a higher wavelength of the solar spectrum in the dye sensitization of TiO2. The organometallic dye, especially (2,2′-bipyridine) ruthenium(II) chloride hexahydrate, attached to TiO2 absorbs sunlight and injects electrons into the conduction band of TiO2, which ultimately promotes CO2 reduction to produce CH4.43 It was also reported that the Perylenediimide derivatives have shown high light harvesting capacity similar to the Ru complex dye.Nguyen et al. found substantial improvements in the photoactivity of metal-doped TiO2–SiO2 mixed oxide-based photocatalysts towards CO2 photoreduction in the presence of H2O and concentrated sunlight in an optical fiber photoreactor (OFPR).44 Fe atoms are introduced into the TiO2–SiO2 lattice during sol–gel process, resulting in the full visible light absorption as well as the effect on product selectivity of the obtained catalyst. Cu–Fe/TiO2 catalyst mainly produced ethylene with the quantum yield of 0.024%, whereas Cu–Fe/TiO2–SiO2 catalyst exhibited favorable methane production with the quantum yield of 0.05%. The overall energy efficiency is found to be higher on Cu–Fe/TiO2–SiO2 (0.018%) than on its Cu–Fe/TiO2 (0.016%). The superior photoactivity of the Cu–Fe/TiO2–SiO2 catalyst under natural sunlight could be ascribed to the efficient charge transfer mechanism between TiO2 and Cu as well as Fe as co-dopants and its full absorption of visible light.
12. Conclusions
In this review, we have focused on the progress in the very stimulating field of the photocatalytic reduction of CO2 with H2O on various types of Ti–oxide catalysts. Upon UV-light irradiation, the reactions on TiO2 powders in the presence of gaseousCO2 and H2O at 275 K produced CH4 as the major product, while on the highly dispersed titanium oxide anchored on porous glass or ordered porous silicas, the formations of CH3OH as well as CH4 were observed as the major products. In situ spectroscopic studies of the system indicated that the photocatalytic reduction of CO2 with H2O is linked to a much higher activity of the charge transfer excited state, i.e., (Ti3+—O−)* of the tetrahedral coordinated Ti–oxide species formed on the surface. Owing to the dramatically growing nature of this research field, we expect that the design strategy described here can offer a helpful overview to the readers in this fascinating area.References
- M. Anpo, H. Yamashita, In Surface Photochemistry; M. Anpo. Ed.; John Wiley & Sons; Chichester, 1996, pp. 117–164 Search PubMed.
- M. Anpo, H. Yamashita, In Heterogeneous Photocatalysts; M. Schiavello, Ed; John Wiley & Sons; Chichester, 1997, pp. 133–168 Search PubMed.
- H. Yamashita, J. Zhang, M. Matsuoka, M. Anpo, In Photofunctional Zeolites; M. Anpo. Ed.; NOVA; New York, 2000, pp. 129–168 Search PubMed.
- M. Anpo, H. Yamashita and S. G. Zhang, Current Opinion in Solid State & Materials Science, 1996, 0, 219–224 Search PubMed.
- M. Anpo, Res. Chem. Intermed., 1989, 9, 67–112 CrossRef.
- M. Anpo, Y. Ichihashi, M. Takeuchi and H. Yamashita, Res. Chem. Intermed., 1998, 24, 143–149 CrossRef CAS.
- H. Yamashita, Y. Ichihashi, M. Takeuchi, S. Kishiguchi and M. Anpo, J. Synchrotron Radiat., 1999, 6, 451–452 CrossRef CAS.
- H. Yamashita, M. Honda, M. Harada, Y. Ichihashi and M. Anpo, J. Phys. Chem. B, 1998, 102, 10707–10711 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–640 CrossRef CAS.
- M. Halmann, In Energy Resources through Photochemistry and Catalysis; M. Grätzel Ed.; Academic Press, New York, 1983, pp. 507–565 Search PubMed.
- M. Anpo and K. Chiba, J. Mol. Catal., 1992, 74, 207–302 CrossRef CAS.
- H. Yamashita, A. Shiga, S. Kawasaki, Y. Ichihashi, S. Ehara and M. Anpo, Energy Convers. Manage., 1995, 36, 617–620 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi and S. Ehara, J. Electroanal. Chem., 1995, 396, 21–26 CrossRef.
- H. Yamashita, H. Nishiguchi, N. Kamada, M. Anpo, Y. Teraoka, H. Hatano, S. Ehara, K. Kikui, L. Palmisano, A. Sclafani, M. A. Fox and M. Schiavello, Res. Chem. Intermed., 1994, 20, 815–823 CrossRef CAS.
- H. Yamashita, N. Kamada, H. He, K. Tanaka, S. Ehara and M. Anpo, Chem. Lett., 1994, 855–858 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Fujii, Y. Ichihashi, S. G. Zhang, D. R. Park, S. Ehara, S. E. Park, J. S. Chang and J. W. Yoo, Stud. Surf. Sci. Catal., 1998, 114, 177–182 CrossRef CAS.
- H. Yamashita, Y. Fuji, Y. Ichihashi, S. G. Zhang, K. Ikeue, D. R. Park, K. Koyano, T. Tatsumi and M. Anpo, Catal. Today, 1998, 45, 221–227 CrossRef CAS.
- M. Anpo, S. G. Zhang, Y. Fujii, Y. Ichihashi, H. Yamashita, K. Koyano and T. Tatsumi, Catal. Today, 1998, 44, 327–332 CrossRef CAS.
- K. Ikeue, H. Yamashita and M. Anpo, Chem. Lett., 1999, 1135–1136 CrossRef CAS.
- H. Yamashita, K. Ikeue, T. Takewaki and M. Anpo, Top. Catal., 2002, 18, 95–100 CrossRef CAS.
- H. Yamashita, S. Kawasaki, Y. Fujii, Y. Ichihashi, S. Ehara, S. E. Park, J. S. Chang, J. W. Yoo and M. Anpo, Stud. Surf. Sci. Catal., 1998, 114, 561–564 CrossRef CAS.
- F. Saladin, L. Forss and I. Kamber, J. Chem. Soc., Chem. Commun., 1995, 533–534 RSC.
- M. Anpo, M. Tomonari and M. A. Fox, J. Phys. Chem., 1989, 93, 7300–7303 CrossRef CAS.
- H. Yamashita, Y. Ichihashi, M. Harada, G. Stewart, M. A. Fox and M. Anpo, J. Catal., 1996, 158, 97–101 CrossRef CAS.
- K. W. Frese, J. Electrochem. Soc., 1991, 138, 3338–3343 CrossRef CAS.
- Slamet, H. W. Nasution, E. Purnama, S. Kosela and J. Gunlazuardi, Catal. Commun., 2005, 6, 313–319 CrossRef CAS.
- J. Raskö and F. Solymosi, J. Phys. Chem., 1994, 98, 7147–7152 CrossRef.
- M. Anpo, M. Matsuoka, Y. Shioya, H. Yamashita, E. Giamello, C. Morterra, M. Che, H. H. Patterson, S. Webber, S. Ouellette and M. A. Fox, J. Phys. Chem., 1994, 98, 5744–5750 CrossRef CAS.
- H. Yamashita, M. Matsuoka, K. Tsuji, Y. Shioya, M. Anpo and M. Che, J. Phys. Chem., 1996, 100, 397–402 CrossRef CAS.
- H. Yamashita, Y. Ichihashi, M. Anpo, M. Hashimoto, C. Louis and M. Che, J. Phys. Chem., 1996, 100, 16041–16044 CrossRef CAS.
- H. Yamashita, S. G. Zhang, Y. Ichihashi, Y. Matsumura, S. Souma, T. Tatsumi and M. Anpo, Appl. Surf. Sci., 1997, 121, 305–309 CrossRef.
- F. Farges, G. E. Brown and J. H. Rehr Jr., Geochim. Cosmochim. Acta, 1996, 60, 3023–3060 CrossRef CAS.
- M. Anpo, N. Aikawa, Y. Kubokawa, M. Che, C. Louis and E. Giamello, J. Phys. Chem., 1985, 89, 5017–5021 CrossRef CAS.
- M. Anpo, N. Aikawa, Y. Kubokawa, M. Che, C. Louis and E. Giamello, J. Phys. Chem., 1985, 89, 5689–5694 CrossRef CAS.
- T. Takewaki, S. J. Hwang, H. Yamashita and M. E. Davis, Microporous Mesoporous Mater., 1999, 32, 265–273 CrossRef CAS.
- M. A. Camblor, A. Corma, P. Esteve, M. Martines and S. Valencia, Chem. Commun., 1997, 795–796 RSC.
- T. Tatsumi and N. Jappar, J. Phys. Chem. B, 1998, 102, 7126–7131 CrossRef CAS.
- W. Lin, H. Han and H. Frei, J. Phys. Chem. B, 2004, 108, 18269–18273 CrossRef CAS.
- K. Ikeue, S. Nozaki, M. Ogawa and M. Anpo, Catal. Lett., 2002, 80, 111–114 CrossRef CAS.
- K. Ikeue, S. Nozaki, M. Ogawa and M. Anpo, Catal. Today, 2002, 74, 241–248 CrossRef CAS.
- H. Yamashita, S. Kawasaki, Y. Ichihashi, M. Harada, M. Anpo, G. Stewart, M. A. Fox, C. Louis and M. Che, J. Phys. Chem. B, 1998, 102, 5870–5875 CrossRef CAS.
- O. Ozcan, F. Yukruk, E. U. Akkaya and D. Uner, Top. Catal., 2007, 44, 523–528 CrossRef CAS.
- T. –H. Nguyen and J. C. S. Wu, Sol. Energy Mater. Sol. Cells, 2008, 92, 864–872 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |