Facile preparation of ordered arrays of polystyrene spheres dissymmetrically decorated with gold nanoparticles at air/liquid interface and their SERS properties†
Yue
Xia
a,
Yingchang
Yang
a,
Jufang
Zheng
b,
Wei
Huang
a and
Zelin
Li
*a
aKey Laboratory of Chemical Biology and Traditional Chinese Medicine Research (Ministry of Education of China), College of Chemistry and Chemical Engineering, Hunan Normal University, Lushan Road, Changsha, 410081, China. E-mail: lizelin@hunnu.edu.cn; Fax: +86 731 88872531; Tel: +86 731 88871533
bKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, Institute of Physical Chemistry, Zhejiang Normal University, Jinhua, 321004, China
First published on 27th March 2012
Abstract
Ordered arrays of polystyrene (PS) spheres dissymmetrically decorated with gold nanoparticles (NPs) were facilely assembled at air/liquid interface by the combination of colloidal crystal templating and modified conventional electroless plating. Distinguishingly, Sn2+ ions served as the reductant and stabilizer in the preparation of Au colloid, and the immersed part of a floating monolayer of pristine PS spheres was also sensitized by the excessive amounts of Sn2+ ions in the Au colloid. Colloidal Au NPs spontaneously aggregated onto the sensitized portion of PS spheres due to oxygen-induced ligand replacement of SnCl3− with Cl−, forming asymmetric PS/Au composite sphere arrays. Moderate heating (60 °C) accelerated the assembly process. The method presented here is convenient, cost-effective and can be extended to prepare asymmetric composite sphere arrays of PS/Pd and PS/Au–Pd. Furthermore, the surface enhanced Raman scattering (SERS) performance of the ordered arrays of asymmetric PS/Au composite spheres was examined.
1. Introduction
Dielectric matrices modified by metallic nanoparticles (NPs) have received continuous attention in the last few years. These materials combine highly desirable size-dependent optical, electrical and catalytic properties of nanoscale metallic particles with dielectric functions of dielectric spheres, showing great potential in advanced functional applications of heterogeneous catalysis,1 photothermal therapy,2 surface-enhanced Raman scattering (SERS),3,4 protein immobilization,5etc. Attracted by the facile synthesis, highly controllable size and easy surface functionalization, polystyrene (PS) or silica (SiO2) dielectric spheres have been widely employed as cores to fabricate dielectric-metal composites with core-shell and hollow structures. In order to effectively bind metallic NPs to relatively inert dielectric spheres, various methods have been suggested to functionalize the surfaces of dielectric spheres, including seeding plating and/or electroless plating,6,7 chemical modification,8,9 layer-by-layer (LBL) assembly,10,11 and combinations of different techniques.12,13 More detailed methods to make Janus particles (or patchy particles) have been reviewed by Kretzschmar et al.14,15 Ordered dielectric-metal sphere arrays can also be obtained by physical vapor deposition,16–18 solution deposition,19,20 electroless deposition21,22 and electrodeposition.23,24 In these cases, pretreatment of dielectric spheres or special techniques and instruments are usually required. Moreover, the fabrication of dielectric-metal films from these techniques was generally performed on a flat solid substrate, making the film transfer difficult. Instead, dielectric-metal composites prepared at gas/liquid interface showed excellent transferability and unsymmetrical features.25,26Tin salts are commonly used in seeding/electroless plating method to fabricate dielectric-metal core-shell spheres, which provide catalytic sites on the whole surface of dielectric spheres to reduce metallic salts forming metallic seeds.6,27 In the following plating method, complete metallic shells are developed on these seeds. In general, the whole surface of dielectric spheres is homogeneously modified by tin salts in solutions, and the following reduction and growth occur in a uniform manner. Thus asymmetric surface modification is hardly obtained using this strategy. We recently demonstrated that Sn2+ ions can serve as a reducing agent and stabilizer to prepare Au colloids.28 Through the oxygen-induced ligand replacement of protective SnCl3− with aggressive Cl−, Au NPs can self-assemble at the air/liquid interface to form a thin film. In such Au colloid, the existence of excess Sn2+ ions can sensitize the surface of dielectric spheres for the assembly of Au NPs. Therefore, ordered arrays of dielectric-metal composites are likely to be obtained by floating assembled monolayers of PS spheres on the surface of SnCl3−-protected Au colloid.
In this paper, a novel colloidal crystal templating method combined with modified conventional electroless plating technique is proposed to prepare ordered arrays of asymmetric PS/Au composite spheres. Taking the air/liquid interface as a dissymmetrization tool, the immersed under-surface of monolayer PS spheres sensitized by Sn2+ acts as a template for the self-assembly of colloidal Au NPs through oxygen-induced ligand replacement, leaving a bare patch on the PS spheres exposed to the air. The process could be accelerated by moderate heating. SERS activities of the prepared PS/Au composite sphere arrays were also investigated by using rhodamine B (RdB) as a probe molecule. Here PS spheres with a diameter of 2 μm were chosen as the template since they can be easily recognized and individually manipulated by conventional optical microscopy.29 Furthermore, similar ordered arrays of asymmetric composite spheres of PS/Pd or PS/Au–Pd can be constructed using the same strategy. The present work reports the following new features: (1) Sn2+ ions play three roles as a reductant, stabilizer and sensitizer; (2) no pretreatment is required to modify PS spheres or Au NPs; (3) the sensitization of PS spheres and the assembly of colloidal Au NPs take place in a one-pot manner; (4) the fabrication of ordered sphere arrays of dielectric-metal composites can be accelerated under moderate temperature; (5) the entire experimental procedure can be easily operated in an ordinary laboratory.
2. Experimental
2.1 Reagents
Monodispersed PS spheres of 2 μm size were purchased from Tianjin Baseline Chromtech Research Center (Tianjin, China). HAuCl4·4H2O and PdCl2 were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Rhodamine B and all other reagents were of analytical grade purity and used directly without further purification, and all solutions were prepared using high-purity water from a Milli-Q water purification system (Millipore Corp., USA).2.2 Preparation of ordered arrays of asymmetric dielectric-metal composite spheres at air/liquid interface
Asymmetric PS/Au composite sphere arrays were prepared according to the following procedure. A monolayer template of PS spheres was assembled as described by Qi et al.26 In brief, 10 μL of 5 wt% PS sphere suspension in water/ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was dropped onto the top surface of a piece of 1 × 1 cm glass substrate that was located at the center of a Petri dish and was surrounded by water. The suspension spread freely on the water surface. Then 10 μL of sodium dodecyl sulfate (2 wt% in water) was dropped onto the water surface, forming a stable monolayer film of PS spheres. The monolayer film of PS spheres was picked up with a silicon plate, and transferred onto the surface of the Au colloid, which were prepared according to the method previously reported by our group28 but with reduced volume of precursor solutions (see Table S1 in ESI†). Then the colloid was kept at room temperature (20 °C) or heated to 60 °C for different periods of time without stirring. Finally, the obtained ordered asymmetric PS/Au composite sphere arrays were transferred onto glass slides or silicon plates, rinsed gently with a solution of 2 M HCl to remove residual soluble tin complexes,28 and followed with high-purity water several times. The cleaned PS/Au composite sphere arrays were dried at room temperature ready for further Raman measurements and morphology characterizations. Asymmetric PS/Pd and PS/Au–Pd sphere arrays were prepared by an identical procedure except for replacing HAuCl4 with PdCl2 or with mixture of PdCl2 and HAuCl4 (see Table S1 in ESI†).
:1) was dropped onto the top surface of a piece of 1 × 1 cm glass substrate that was located at the center of a Petri dish and was surrounded by water. The suspension spread freely on the water surface. Then 10 μL of sodium dodecyl sulfate (2 wt% in water) was dropped onto the water surface, forming a stable monolayer film of PS spheres. The monolayer film of PS spheres was picked up with a silicon plate, and transferred onto the surface of the Au colloid, which were prepared according to the method previously reported by our group28 but with reduced volume of precursor solutions (see Table S1 in ESI†). Then the colloid was kept at room temperature (20 °C) or heated to 60 °C for different periods of time without stirring. Finally, the obtained ordered asymmetric PS/Au composite sphere arrays were transferred onto glass slides or silicon plates, rinsed gently with a solution of 2 M HCl to remove residual soluble tin complexes,28 and followed with high-purity water several times. The cleaned PS/Au composite sphere arrays were dried at room temperature ready for further Raman measurements and morphology characterizations. Asymmetric PS/Pd and PS/Au–Pd sphere arrays were prepared by an identical procedure except for replacing HAuCl4 with PdCl2 or with mixture of PdCl2 and HAuCl4 (see Table S1 in ESI†).
2.3 Characterizations
The morphologies and surface component of dielectric-metal composite sphere arrays were characterized by a Hitachi S-4800 field emission scanning electron microscope (SEM) combining with energy-dispersive X-ray (EDX) spectroscopy. Though PS spheres were dielectric particles, additional Au sputtering coating usually recommended for high resolution SEM images was not performed in order to obtain the exact surface component. Raman spectra were obtained using a Renishaw RM1000 confocal Raman spectrometer (Renishaw, U.K.). The prepared PS/Au composite sphere arrays loaded on glass slides were dipped in 1 μM RdB for 4 h, washed thoroughly with high-purity water and dried at room temperature while being protected from light. A 633 nm He–Ne laser was used as the excitation source with a 1% operating power (6.5 mW at 100%) to avoid the photodegradation of RdB. The Raman data were an averaged value of five replicate measurements collected from different surface areas with an acquisition time of 10 s for each sample.3. Results and discussion
3.1 Fabrication principle of ordered arrays of asymmetric PS/Au composite spheres at air/liquid interface
The fabrication principle of an asymmetric PS/Au composite sphere array is schematically depicted in Fig. 1. A monolayer of PS spheres obtained according to Qi et al.26 was transferred onto the surface of Au colloid that was produced from the reduction of HAuCl4 solution by excess Sn2+ ions (Fig. 1A). The considerable amounts of remaining Sn2+ ions existed in the form of complex ions SnCl3− could stabilize the Au colloid.28 Both SnCl3− and SnCl62– in the Au colloid would adsorb onto the under-surface of the PS spheres.30 Because commercially available PS spheres often contain terminal hydroxyl and carboxylate groups on the sphere exterior,31 reactions of the complex ions SnCl3− and SnCl62– in the colloid with carboxylated or hydroxylated PS sphere surface are assumed to occur,30,32 forming partially Sn-functionalized PS spheres (Fig. 1B). This process is equivalent to the so-called “pretreatment steps” in electroless plating.6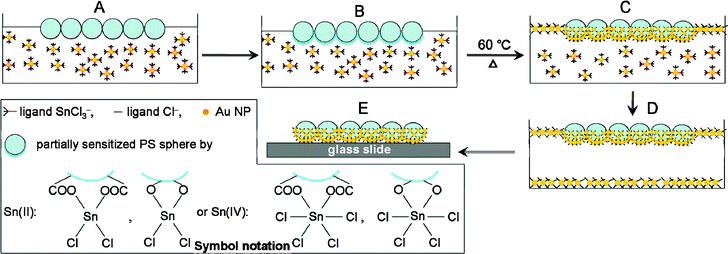 | ||
| Fig. 1 Schematic illustration for the preparation of ordered arrays of asymmetric PS/Au composite spheres at air/liquid interface. (A) A monolayer of PS spheres was spread over the Au colloid surface as a template, (B) unreacted Sn2+ ions in the Au colloid sensitized the immersed region of PS spheres, (C) colloidal Au NPs were speedily self-assembled at the air/liquid interface and on the sensitized portions of PS spheres by heating the Au colloid, (D) no more colloidal Au NPs could be supplied for further assembly when Au colloid became clear, and (E) the ordered array of asymmetric PS/Au composite spheres was transferred on a glass slide or a silicon plate for further experiments. | ||
As reported previously,28 the protective SnCl3− ligands on the Au NPs near the air/liquid interface were replaced with the aggressive Cl− by the oxygen-induced ligand replacement, thus the unstable Au NPs began to aggregate at air/liquid interface, where the partially Sn-sensitized PS spheres served as template for the assembly of Au NPs with Cl− ligands. Moderate heating (60 °C) can accelerate these processes. Considering that heating could also induce the aggregation of colloidal NPs at air/liquid interface,33 we heated the SnCl3−-protected Au colloid at 60 °C under N2 atmosphere to avoid the oxygen-induced ligand replacement. However, no assembled Au film was observed at the gas/liquid interface under N2 atmosphere (Fig. S1, ESI†). Therefore, it can be concluded that the aggregation of Au NPs at air/liquid interface under heating mainly resulted from the oxygen-induced ligand replacement. We speculated that the Au NPs with Cl− ligands could link to the partially Sn-sensitized PS spheres through bridged coordinate bond like Au–Cl–Sn.34 Finally, the immersed portion of the PS spheres was almost completely coated with Au NPs, leaving a bare patch exposed to the air. We must point out here that the Au colloids exposed to the air were unstable, so some colloidal Au NPs self-assembled at the air/liquid interface without PS spheres, or were deposited onto the bottom of weighing bottle.28 Finally, the red brown Au colloid became clear, implying that no Au NPs could be supplied for further interface assembly (Fig. 1D). The prepared film of asymmetric PS/Au composite sphere array can be transferred on a glass slide or a silicon plate for further experiments (Fig. 1E).
3.2 Effects of temperature and heating time on the fabrication of ordered arrays of asymmetric PS/Au composite spheres
PS spheres with tunable coverage of assembled Au NPs were easily obtained by controlling temperature and thermal treatment time. It took at least 2 h at room temperature (20 °C) to form a thin film of aggregated Au NPs at the air/liquid interface,28 but only needed 20 min or so at 60 °C because heating can accelerate the assembly process. Fig. 2(a–e) demonstrated the morphological evolution of the PS/Au composite spheres at 60 °C. In the initial 5 min, a small amount of Au NPs were assembled on the PS spheres in cycle locating at the air/liquid interface, while no Au NPs were found in the lower immersion portion of the PS spheres (Fig. 2a), indicating that it was the species Au NPs–Cl− (instead of Au NPs–SnCl3−) that adsorbed on PS spheres. With the oxidation consumption of SnCl3− at the air/liquid interface by oxygen from the air, the oxygen-induced ligand replacement of SnCl3− with Cl− developed deeper into the colloid. So, more and more Au NPs were attached onto the immersion portion of the PS spheres from 10 min (Fig. 2b) to 40 min (Fig. 2e). Here, the partially sensitized PS spheres acted as template for the assembly of Au NPs. Liz-Marzán and co-workers6 reported a similar method to deposit Ag NPs on the whole SiO2 sphere surface by a repeated two-step method. First, Sn2+ adsorbed on the surface of SiO2 spheres dispersed in a solution of SnCl2, and then the Sn2+-sensitized SiO2 spheres were separated from the SnCl2 solution and mixed with a AgNO3 solution. By repeating these two steps, they obtained uniformly distributed Ag NPs on whole SiO2 sphere surfaces. In this work, we integrated the two-step electroless plating processes into one and fabricated ordered arrays of asymmetric PS/Au composite spheres at air/liquid interface. Note that each asymmetric PS/Au composite sphere in Fig. 2 has a well-defined dark area where no Au NPs were present, corresponding to the region of PS spheres exposed to the air. The low magnification SEM image of Fig. 2f indicated that our strategy can be used to create a large-scaled asymmetric PS/Au composite sphere array at air/liquid interface with a highly local ordered arrangement (the inset of Fig. 2g). The above SEM images indicated that the bridged coordinate bond Au–Cl–Sn between the partially Sn-sensitized PS spheres and Au NPs was strong enough to survive rinsing. EDX analysis confirmed the existence of Au and C elements (Fig. 2g). Since a monolayer or so of Sn was present on the PS spheres (Fig. 1), it cannot be detected by EDX due to its low sensitivity. Higher temperature than 60 °C could accelerate the formation of Au film. However, PS spheres may wobble or rotate at the air/liquid interface induced by thermal disturbance, rendering partial modification of PS spheres impossible. | ||
| Fig. 2 Detailed SEM images of PS/Au composite spheres assembled at 60 °C for (a) 5 min, (b) 10 min, (c) 20 min, (d) 30 min and (e) 40 min. (f) A large-scaled morphology of (e), and (g) EDX patterns of (f). The inset in (g) shows a highly ordered layer of PS/Au composite spheres in (f). | ||
For comparison, the morphological evolution of asymmetric PS/Au composite spheres assembled at room temperature (20 °C) are shown in Fig. 3. At relatively low temperature, the rate of oxygen-induced ligand replacement of protective SnCl3− with aggressive Cl− was rather slow,28 and much longer time (at least ~2 h) was required to decorate the immersed region of PS spheres with Au NPs (Fig. 3a). The Au NPs attached onto the PS sphere became larger around 6 h (Fig. 3b), and finally formed nano-network-like structures (Fig. 3c) due to coalescence of Au NPs during the long aging time in the strong electrolyte of HCl. This situation was not observed at 60 °C in Fig. 2e due to the faster assembly of Au NPs in short time. Fig. 3d displayed a large-scaled morphology of Fig. 3c. Several random oriented PS/Au composite spheres on Si substrate were exhibited in Fig. 3e, including tilted and upturned ones. The EDX spectrum of PS/Au composite spheres in Fig. 3f showed the presence of Au and C elements, and the signal of Si was from the Si substrate.
 | ||
| Fig. 3 Detailed SEM images of PS/Au composite spheres assembled at room temperature (20 °C) for (a) 2 h, (b) 6 h and (c) 12 h. (d) A large-scaled morphology of (c). (e) Several random oriented spheres on Si substrate. (f) The EDX spectrum of (d). | ||
In order to prove that Sn2+ ions are essential in the fabrication of such PS/Au composite spheres, we prepared another Au colloid without Sn2+ ions using the procedure developed by Frens35 to replace the Sn2+-protected Au colloid. We previously found that spontaneous self-assembly of Au NPs protected by citrate occurred at the gas/liquid interface under the vapor of formic acid.36 However, in the presence of monolayer PS spheres floating on the citrate-protected Au colloid, Au NPs were scarcely attached on the surface of PS spheres as displayed in Fig. S2 in the ESI†.
3.3 Fabrication of ordered arrays of asymmetric PS/Pd and PS/Au–Pd composite spheres at the air/liquid interface
In electroless plating, an acid solution of PdCl2 usually mixed with SnCl2 as a composite PdCl2/SnCl2 sensitizer to activate non-conductive surfaces for electroless metal deposition.37–41 Recent evidence indicated that the Pd–Sn colloidal sensitizer from mixed PdCl2/SnCl2 solution consisted of NPs with a Pd–Sn alloy core and a Sn shell.42 We found that no self-assembled film of Pd NPs appeared at the air/liquid interface unless pre-heated PdCl2 solution and SnCl2 solution respectively were used and mixed together.43 In this work, we heated the mixed PdCl2/SnCl2 solution at 60 °C for 5 h until the solution became clear, and ordered arrays of PS/Pd composite spheres were obtained (Fig. 4a and 4b). EDX analysis in Fig. 4d indicated the presence of Pd and C elements, and the signal of Si was from the Si substrate. Though the spent time (5 h) in the preparation of PS/Pd composite spheres was much longer than that in the preparation of S/Au composite spheres (40 min), tiny Pd NPs instead of large Pd NPs were observed on PS sphere surfaces, which were similar to the Au NPs assembled by heat treatment at 60 °C for 20 min (Fig. 2b). At higher temperature (90 °C), the self-assembly of Pd NPs at air/liquid interface finished in shorter time (15 min), and larger Pd NPs were observed on PS sphere surfaces (Fig. 4c, inset). However, the whole surfaces of PS spheres were nearly fully coated with Pd NPs due to the intense thermal disturbance (Fig. 4c). | ||
| Fig. 4 SEM images of PS/Pd composite spheres assembled (a, b) at 60 °C for 5 h and (c) at 90 °C for 15 min. (d) The EDX spectrum of (a). | ||
We found that the addition of PdCl2 into HAuCl4 solution was beneficial to the fabrication of composite spheres with more compact metallic coatings. We first prepared mixed solutions of PdCl2, HAuCl4 and SnCl2 with varied Pd/Au ratio (as shown in Table S1 in the ESI†), and then transferred the monolayer PS sphere template onto the mixed solution surface. By heating the mixed solution at 60 °C for a given time, a continuous cup-shaped shell of assembled tiny metallic NPs was observed on the PS spheres (Fig. 5). The morphology is different from the scattered Au NPs and nano-network structures shown in Fig. 2e and Fig. 3c, respectively. Such results may occur from the enhanced sensitizing effect of Pd species as they performed in electroless plating. It was found that the time spent for the self-assembly of metallic NPs at the air/liquid interface was dependent on the precursor ratios of PdCl2 and HAuCl4 at fixed temperature (60 °C). Higher content of Pd2+ ions in the mixed solutions resulted in longer time to obtain dielectric-metal composite spheres at air/liquid interface (see the note of Fig. 5). It is understandable since much longer time was required to prepare PS/Pd composite spheres at 60 °C (Fig. 4). The solutions became clear at the end of assembly. These results indicate that the colloids were more stable in the presence of PdCl2. The EDX spectra in Fig. 5 reveal the presence of Au, Pd and C elements.
 | ||
| Fig. 5 SEM images of asymmetric PS/Au–Pd composite spheres prepared at 60 °C with different molar ratios of PdCl2 to HAuCl4 in the mixed solutions for different thermal treatment time: (a1, b1) 1:3.6 for 1.5 h, (a2, b2) 1:1.2 for 3 h, and (c1, c2) the corresponding EDX spectra. | ||
3.4 SERS properties of ordered arrays of PS/Au composite spheres
Taking RdB as a model analyte, we investigated the SERS performances of PS/Au composite sphere arrays. With the help of Raman microscopy, it was quite easy to find an ordered area of composite spheres (the inset of Fig. 6). Fig. 6 displays the SERS spectra of RdB on PS/Au composite sphere arrays obtained at 60 °C. We found that SERS activity was strongly dependent on the aggregation state of Au NPs. The SERS spectrum of RdB became stronger with the thermal treatment time from 20 to 40 min for the PS/Au composite sphere arrays (Fig. 6a–c) and the most characteristic peaks of RdB molecules44 at 1197, 1358, 1510, 1592 and 1648 cm−1 were clearly observed in Fig. 6c. The increased number and size of Au NPs on the PS sphere surface (Fig. 2a–c) account for the signal enhancement of RdB with longer thermal treatment time.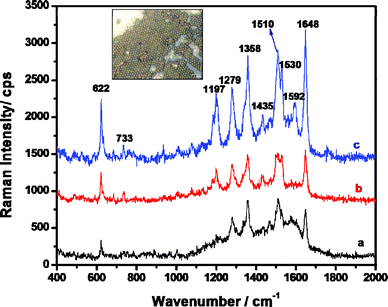 | ||
| Fig. 6 SERS spectra of RdB on the ordered arrays of asymmetric PS/Au composite spheres prepared at 60 °C with different thermal treatment time: (a) 20 min, (b) 30 min and (c) 40 min. Inset: a typical optical micrograph of PS/Au composite sphere arrays observed by Raman microscopy. Proper offset has been made to see the spectra clearly. | ||
The SERS properties of PS/Au composite sphere arrays prepared at room temperature (20 °C) were also examined (Fig. 7). The SERS intensities of RdB increased (Fig. 7a–c) with prolonged assembly time due to the increased number and size of Au NPs on PS spheres (Fig. 3a–c). The adsorbed RdB showed much stronger signals on the PS/Au composite sphere array with nano-network-like aggregated Au NPs produced at room temperature (Fig. 3c) than on PS/Au composite sphere arrays formed at 60 °C (Fig. 2e). Both experimental and theoretical results have demonstrated that strong electromagnetic field enhancement will be generated at crevices or junctions of particle aggregates,45–47 and here the “hot spots” of SERS mainly resulted from the closely packed Au NPs assembled on the PS spheres with nano-network structures48 and the aggregation of PS/Au spheres.
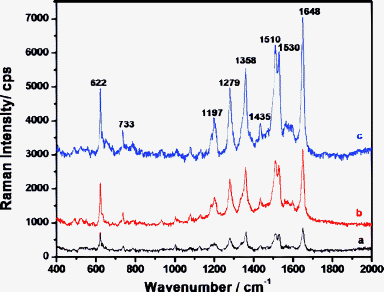 | ||
| Fig. 7 SERS spectra of RdB on the ordered arrays of PS/Au composite spheres prepared at room temperature (20 °C) with different reaction time: (a) 2 h, (b) 6 h and (c) 12 h. Proper offset has been made to see the spectra clearly. | ||
For comparison, SERS spectra of RdB on aggregated Au NPs without PS spheres, on a Au electrode roughened by electrochemical oxidation-reduction cycles (ORC),49 and on the PS/Au composite sphere array are exhibited in Fig. S3 in ESI†. We found that the SERS signals of RdB changed greatly on the aggregated Au NPs without PS spheres, comparing spectra 1 and 3 that were taken at regions A and B respectively (Fig. S3 in ESI†). However, PS/Au composite sphere arrays exhibited rather homogenous SERS signals. In addition, the PS/Au composite sphere array also showed stronger SERS activity (Fig. S3a†, spectrum 2) than that on the aggregated Au NPs loaded on a flat substrate (Fig. S3a†, spectra 1 and 3) and on the rough Au electrode prepared by the electrochemical ORC method (Fig. S3a†, spectrum 4). One reason was attributed to the effectively dispersed Au NPs on PS spheres where a lot of SERS-active nanogaps were formed. Another reason could be ascribed to the curvature of PS spheres which may have certain effect on focusing light. Lin et al.50 investigated the enhanced Raman scattering by assembling PS microspheres on Au single crystal surface. They suggested that when a laser was focused on the PS micorspheres with an appropriate diameter, nanojets occurred which led to the highly localised electromagnetic field and the enhanced Raman signals. Here, similar nanojets would be formed because the laser could pass through the top of asymmetric PS/Au microspheres. Further studies should be conducted to exploit the application of the asymmetric PS/Au structures in SERS.
4. Conclusions
In summary, we have demonstrated a new and efficient route to prepare ordered arrays of asymmetric PS/Au composite spheres at the air/liquid interface of Sn2+-stabilized Au colloids. Here Sn2+ ions played three roles during the preparation process: (i) reductant to produce Au NPs from HAuCl4 solution, (ii) stabilizer to protect Au NPs in the form of SnCl3−, and (iii) sensitizer to functionalize PS spheres. The partially Sn2+-sensitized PS spheres acted as a template for the assembly of Au NPs driven by the oxygen-induced ligand replacement. The assembly process can be accelerated by moderate heating. The adsorbed amounts of Au NPs on the surface of partially immersed PS spheres were tunable by controlling the temperature and heating time. This method can be extended to fabricate ordered arrays of PS/Pd and PS/Au–Pd composite spheres. The SERS properties of PS/Au composite sphere arrays were also investigated. It was found that the SERS activity of PS/Au composite sphere arrays was mainly dependent on the aggregation state of Au NPs on PS spheres. Local electromagnetic field enhancement from the closely packed Au NPs assembled on the PS spheres and from the aggregation of PS/Au spheres was believed to be responsible for the strong SERS signals. Comparing with random aggregated Au NPs, the ordered array of PS/Au composite spheres showed homogeneity and reproducibility in their local structure, which was hoped to be a potential substrate for quantitative SERS analysis. PS/Au composite spheres also showed high SERS activity in comparison with aggregated Au NPs without PS microspheres and with rough Au electrode prepared by electrochemical ORC method. Further work should be done to exploit the application of the asymmetric PS/Au composite sphere array in SERS.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (Grant Nos. 21173075 and 21003045), Ph.D. Programs Foundation of the Education Ministry of China (Grant No. 20104306110003), Natural Science Foundation of Zhejiang Province of China (Grant No. Y4090658), the Open Foundation of Key Laboratory of the Ministry of Education for Advanced Catalysis Materials & Zhejiang Key Laboratory for Reactive Chemistry on Solid Surfaces (Grant No. DH201001), and Aid Program for Science and Technology Innovative Research Team in Higher Educational Institutions of Hunan Province.References
- C. W. Chen, T. Serizawa and M. Akashi, Chem. Mater., 1999, 11, 1381–1389 CrossRef CAS.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef CAS.
- M. Spuch-Calvar, L. Rodríguez-Lorenzo, M. P. Morales, R. A. Álvarez-Puebla and L. M. Liz-Marzán, J. Phys. Chem. C, 2009, 113, 3373–3377 CAS.
- C. Farcau and S. Astilean, J. Phys. Chem. C, 2011, 114, 11717–11722 Search PubMed.
- Y. C. Cao, Z. Wang, X. Jin, X. F. Hua, M. X. Liu and Y. D. Zhao, Colloids Surf., A, 2009, 334, 53–58 CrossRef CAS.
- Y. Kobayashi, V. Salgueiriño-Maceira and L. M. Liz-Marzán, Chem. Mater., 2001, 13, 1630–1633 CrossRef CAS.
- J. B. Liu, W. Dong, P. Zhang, S. Z. Wang, J. H. Zhang and Z. L. Wang, Langmuir, 2005, 21, 1683–1686 CrossRef CAS.
- T. Pham, J. B. Jackson, N. J. Halas and T. R. Lee, Langmuir, 2002, 18, 4915–4920 CrossRef CAS.
- B. Sadtler and A. Wei, Chem. Commun., 2002, 15, 1604–1605 RSC.
- Z. Liang, A. S. Susha and F. Caruso, Adv. Mater., 2002, 14, 1160–1164 CrossRef CAS.
- Z. Liang, A. Susha and F. Caruso, Chem. Mater., 2003, 15, 3176–3183 CrossRef CAS.
- T. Ji, V. G. Lirtsman, Y. Avny and D. Davidov, Adv. Mater., 2001, 13, 1253–1256 CrossRef CAS.
- Z. Chen, Z. L. Wang, P. Zhan, J. H. Zhang, W. Y. Zhang, H. T. Wang and N. B. Ming, Langmuir, 2004, 20, 3042–3046 CrossRef CAS.
- I. Kretzschmar and J. H. K. Song, Curr. Opin. Colloid Interface Sci., 2011, 16, 84–95 CrossRef CAS.
- A. B. Pawar and I. Kretzschmar, Macromol. Rapid Commun., 2010, 31, 150–168 CrossRef CAS.
- P. Jiang and M. J. McFarland, J. Am. Chem. Soc., 2005, 127, 3710–3711 CrossRef CAS.
- D. S. Raimundo, A. C. Sant'Ana, F. J. R. Fernandez and W. J. Salcedo, Phys. Status Solidi A, 2007, 204, 973–978 CrossRef CAS.
- M. Xu, N. Lu, H. Xu, D. Qi, Y. Wang and L. Chi, Langmuir, 2009, 25, 11216–11220 CrossRef CAS.
- S. Kubo, Z. Z. Gu, D. A. Tryk, Y. Ohko, O. Sato and A. Fujishima, Langmuir, 2002, 18, 5043–5046 CrossRef CAS.
- F. Sun, W. Cai, Y. Li, B. Cao, Y. Lei and L. Zhang, Adv. Funct. Mater., 2004, 14, 283–288 CrossRef CAS.
- P. Jiang, J. Cizeron, J. F. Bertone and V. L. Colvin, J. Am. Chem. Soc., 1999, 121, 7957–7958 CrossRef CAS.
- L. Lu, I. Randjelovic, R. Capek, N. Gaponik, J. Yang, H. Zhang and A. Eychmüller, Chem. Mater., 2005, 17, 5731–5736 CrossRef CAS.
- F. Sun, W. Cai, Y. Li, B. Cao, F. Lu, G. Duan and L. Zhang, Adv. Mater., 2004, 16, 1116–1121 CrossRef CAS.
- P. N. Bartlett, J. J. Baumberg, S. Coyle and M. E. Abdelsalam, Faraday Discuss., 2004, 125, 117–132 RSC.
- L. Petit, J.-P. Manaud, C. Mingotaud, S. Ravaine and E. Duguet, Mater. Lett., 2001, 51, 478–484 CrossRef CAS.
- C. Li, G. Hong and L. Qi, Chem. Mater., 2010, 22, 476–481 CrossRef CAS.
- M. Schierhorn and L. M. Liz-Marzán, Nano Lett., 2002, 2, 13–16 CrossRef CAS.
- M. Wang, S. Chen, Y. Xia, Y. Zhang, W. Huang, J. Zheng and Z. Li, Langmuir, 2010, 26, 9351–9356 CrossRef CAS.
- L. Piao, S. Park, H. B. Lee, K. Kim, J. Kim and T. D. Chung, Anal. Chem., 2010, 82, 447–451 CrossRef CAS.
- L. R. Pederson, Sol. Energy Mater., 1982, 6, 221–232 CrossRef CAS.
- L. Johnson and D. A. Walsh, J. Mater. Chem., 2011, 21, 7555–7558 RSC.
- M. Bhagwat, P. Shah and V. Ramaswamy, Mater. Lett., 2003, 57, 1604–1611 CrossRef CAS.
- Y. Jin and S. Dong, Angew. Chem., Int. Ed., 2002, 41, 1040–1044 CrossRef CAS.
- V. I. Shiryaev and Y. F. Mironov, Russ. Chem. Rev., 1983, 52, 184–200 CrossRef.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CAS.
- Y. R. Zhang, Y. Z. Xu, Y. Xia, W. Huang, F. A. Liu, Y. C. Yang and Z. L. Li, J. Colloid Interface Sci., 2011, 359, 536–541 CrossRef CAS.
- R. L. Cohen and K. W. West, J. Electrochem. Soc., 1973, 120, 502–508 CrossRef CAS.
- R. L. Cohen and R. L. Meek, J. Colloid Interface Sci., 1976, 55, 156–162 CrossRef CAS.
- N. Feldstein, M. Schlesinger, N. E. Hedgecock and S. L. Chow, J. Electrochem. Soc., 1974, 121, 738–744 CrossRef CAS.
- R. L. Meek, J. Electrochem. Soc., 1975, 122, 1177–1185 CrossRef CAS.
- M. Froment, E. Queau, J. R. Martin and G. Stremsdoerfer, J. Electrochem. Soc., 1995, 142, 3373–3377 CrossRef CAS.
- O. Holderer, T. Epicier, C. Esnouf and G. Fuchs, J. Phys. Chem. B, 2003, 107, 1723–1726 CrossRef CAS.
- Y. R. Zhang, M. H. Wang, J. F. Zheng and Z. L. Li, Int. J. Electrochem. Sci., 2010, 5, 1605–1611 CAS.
- D. Li, D. Li, Y. Li, J. S. Fossey and Y. Long, J. Mater. Chem., 2010, 20, 3688–3693 RSC.
- F. J. García-Vidal and J. B. Pendry, Phys. Rev. Lett., 1996, 77, 1163–1166 CrossRef.
- K. Kim, D. Shin, K. L. Kim and K. S. Shin, Phys. Chem. Chem. Phys., 2010, 12, 3747–3752 RSC.
- K. L. Wustholz, A.-I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. V. Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS.
- Y. Yang, Y. Xia, W. Huang, J. Zheng and Z. Li, J. Solid State Electrochem., 2012, 16, 1733 CrossRef CAS.
- P. Gao, D. Gosztola, L.-W. H. Leung and M. J. Weaver, J. Electroanal. Chem., 1987, 233, 211–222 CrossRef CAS.
- X. Lin, X. Wang, Z. Liu and B. Ren, Acta Phys.-Chim. Sin., 2008, 24, 1941–1944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis conditions of PS/Au, PS/Pd and PS/Au–Pd composite spheres; a photograph for the SnCl3−- protected Au colloid after being heated at 60 °C for 40 min under N2 atmosphere; a SEM image of PS spheres floating on the surface of citrate-protected Au colloid after contacting formic acid vapor for 6 h; and SERS comparison on different substrates. See DOI: 10.1039/c2ra01301k |
| This journal is © The Royal Society of Chemistry 2012 |