DOI:
10.1039/C2RA01293F
(Paper)
RSC Adv., 2012,
2, 2568-2575
Collision-induced dissociation of singly and doubly charged CuII–cytidine complexes in the gas phase: an experimental and computational study†
Received
13th December 2011
, Accepted 14th December 2011
First published on 14th December 2011
Abstract
The complexes of doubly charged cations, [CuLn]2+, [CuL(MeOH)n]2+ and single charged cations, [Cu(L–H)Ln]+, [L+H]+, [L2+H]+, and [B+H]+, etc. (L = cytidine, B = cytosine) were observed via electrospray of water and methanol solutions of Cu(NO3)2 and cytidine. The peak distribution for [CuLn]2+ shows that the [CuL2]2+ and [CuL4]2+ are the highest peaks, then followed by [CuL3]2+, [CuL5]2+, etc.Collision-induced dissociation (CID) of [Cu(L–H)Ln]+(n = 0,1), [CuLn]2+(n = 2–6) and [CuL(MeOH)n]2+(n = 1, 2) was investigated using the ESI-MS/MS instrument, and the structure and thermal energy for the most of these complexes were also calculated with ab initio and DFT methods through Gaussian 09 program. For [Cu(L–H)L0-1]+, the primary dissociation only involved a ribose group, including ribose group lost, ribose side chain and ribose ring broken. The calculation result shows that the deprotonated site for [Cu(L–H)]+ is the N-atom in the amino group of the cytidine base. The inter-ligand proton transfer was observed to dominate the CID of all doubly charged CuII–cytidine complexes, [CuLn]2+ (n = 2–6), and other channels, including charge reduction dissociation, lost neutral ligands were also observed. The protonated cytidine dimer, [L2+H]+, produced in the CID of [CuLn]2+ (n = 4–6), can further break into [L+ H]+ and neutral L, and the structure is proposed to be from the interaction of two cytidinesviahydrogen bonds. The protonated trimeric cytidine was also observed in the dissociation of [CuL6]2+. Three charge reduction dissociation channels were observed in the CID of [CuL(MeOH)]2+. Unlike the CuII–guanosine complex, the radical cation [CuLn]+ was not observed in the CID process of the [CuLn]2+, and the reasons are attributed to the higher ionization energy and proton affinity, which makes the inter-ligand proton transfer more favorable than the electron transfer from ligand to Cu2+.
1. Introduction
There is considerable interest in the interaction of metal cations with nucleosides in view of speculation that metal cations play a crucial role in gene expression (replication, transcription and translation),1–5 in particular the multiple charged cations, because many metals of chemical and biochemical importance commonly occur in their higher oxidation states.6–8 In the liquid-phase, the stability and coordination chemistry of metal cations, especially the multiply charged cations, to nucleosides and nucleotides have been studied quite extensively using NMR, X-ray and other technologies.9–18 But in the gas-phase, there are challenges to generate the multiply charged ion–solvent complex for easy charge transfer from the solvent to metal ions. In the 1990s, Kebarle et al. have demonstrated that electrospray could be used with considerable success to generate a range of solvated doubly and triply charged transition metal cations.19–22 Since then, electrospray ionization (ESI) mass spectrometry (MS) has become the most frequently used technique for the study of the gas-phase complex of monovalent and divalent metal cations with a variety of protic and aprotic ligands, including water,23–27alcohol,28–30acetone and other ketones,23,28,31acetonile,29,32pyridine,19,29dimethysulfoxide,20,22,28 and some DNA bases,33–38etc. With the development of mass spectrometry, the computation and/or MS has proved to be very useful to study the interaction of metal cations with ligands. Very recently, Marino et al.39 have studied the theoretical interaction of rubidium and cesium mono-, strontium and barium bi-cations with DNA and RNA bases; Niu et al.40 studied the interaction of sulfur- and aminopyridine-containing chelating resins with Hg(II) and Pb(II) through calculation; Dunbar et al.41 observed the encapsulation of metal cations by the PhePhe ligand through ESI-MS and DFT calculations.
The biologically important d9 open shell Cu2+ cation has rich redox chemistry and is closely associated with DNA bases.42–45 The activity is connected with a relatively high electron affinity and the CuII oxidation state could easily reduce to CuI. From this point, it is quite necessary to understand the interaction of Cu2+ with the nucleoside in order to study the influence of the Cu2+ in the DNA damage process. Recently , the interactions of Cu2+ with guanosine were investigated, and the radical cations of monomeric and dimeric molecular guansoine were generated by the dissociation of the doubly charged CuII–guanosine complexes.46 Now the study of gas-phase copper complexes using MS has grown into an active and promising research area.47–52
Cytidine (L) is one of the main nucleosides which occur in RNA or DNA, where it participates in stacking interactions and hydrogen bonding to preserve and transfer the genetic information.4,6 Most of the investigations about the interaction of metal caitons with cytidine were done in the liquid-phase.53–55 Recently, a report about the interaction of cytidine with metal cations measured by ESI-MS showed that two types of cytidine clustering depending on coexisting ions were observed. One was the electrostatic interaction, and the other was the coordination interaction.34,56 Almost at the same time, the guanosine–cytidine dinucleoside that self-assembles into a trimetric supramolecule was presented.57
The exposure of DNA to Cu2+ can have different consequences such as single and double strand cleavage, base modification, and formation of abasic sites. So the nature of the interactions of Cu2+ with nucleoside bases should be understood in order to unravel the mechanism for such consequences. 58,59 In this study, we report the gas phase interaction of the Cu2+ with cytidine by ESI mass spectrometry and calculation. Singly and doubly charged complexes of CuII–cytidine were formed, and varied fragmentation chemistries, including inter-ligand proton transfer, ligand elimination, ligand dissociation charge reduction, etc. were observed for the CID of CuII–cytidine complexes. The structure, thermal energy and entropy for most of these CuII–cytidine complexes and their fragmentation products were calculated with ab initio and DFT theory through Gaussian 09 program.60
2. Methods
Experiments were performed using API 2000 (AB Sciex) mass spectrometer equipped with an ESI source. The operating conditions were as follows: ESI positive ion mode; ion spray voltage, 5500 V; ring-electrode potential, 300 V; varied potential values between the orifice and the skimmer; curtain gas flow rate, 10 arbitrary instrument units of N2; nebulizer gas flow rate, 20 arbitrary instrument units dry air; and sample flow rate, 5 μL min−1.
MS/MS was carried out in the product ion monitoring modes with N2 as the collision gas with a flow rate about 9 arbitrary units. The first quadrupole (Q1) was used to select the precursor ions, and the collision induced dissociation performed in the second quadrupole (Q2), the product ions were mass analyzed via the third quadrupole (Q3). The collision energy (Elab) was typically 10–50 ev.
Cu2+ was generated from its nitrate hydrate (Aldrich, p.a 99.99%) without further purification; cytidine was purchased from SIGMA p.a. 98%. HPLC degree methanol and Millipore (18.2 mΩ) water were used to prepare the solvent mixture. Cytidine and Cu2+ metal salt were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 water–methanol mixture.
:3 water–methanol mixture.
2.2 Theoretical method
Calculations were carried out using both the ab initio method and the B3LYP (Becke, three-parameter, Lee–Yang–Parr)/density functional theory (DFT) method. These methods were implemented in the Gaussian 09 program package.60 The UNIX version with standard parameters of this package was installed on the computing cluster in Shanghai University. DFT methods provide an adequate compromise between the desired chemical accuracy and heavy demands on computer time and power. Moreover, DFT methods and in particular B3LYP have been used satisfactorily in many studies of drug design, as well as in other nucleosides and nucleic bases, and they predict vibrational frequencies better than Hartree–Fock (HF) and MP2 methods.61,62 It was well-established that weak interactions were poorly described by DFT methods. However, the hydration of uracil and their derivatives had been reasonably well modeled by DFT. Standard density functional theory calculations were performed using the Gaussian09 suite of programs. Geometries were optimized using Becke's hybrid B3LYP functional and the 6-311+G(d) basis set on C, H, N and O. The copper electrons were handled by the Hay–Wadt effective core potential (ECP) with the LANL2DZ basis set.63 Spin-unrestricted calculations were used for all open-shell systems. The calculated frequencies were used to calculate zero-point energies and thermal enthalpy corrections. Improved energies were obtained by single point calculations with B3LYP using the larger 6-311+G(d) basis set on all atoms or in combination with ECP/LANL2DZ on Cu. Atomic spin and charge densities were calculated using natural population analysis.
3. Results and discussion
Complexes of the type [CuLn]2+, [CuL(MeOH)n]2+ and [Cu(L–H)]+, etc. were readily observed by electrospray of a solution containing Cu2+ and cytidine (L) molecules. The value n depends on the concentration ratio of Cu2+ to cytidine and working in conditions where the declustering potential (DP) is high or low. Keeping the concentration ratio unchanged, if the DP is high, above 50 volts, the maximum value n will reduce, and dissociation charge reduction and other dissociation channels will take place in the source, and lead to diversified MS products. For these reasons, in this experiment we usually work at low DP so as to get the reasonably high abundance of Cu(II)-cytidine complexes. High DP is just used for further CID of the second and higher order products from primary complex dissociation.
The ESI mass spectra of cytidine in the presence of Cu2+ at different concentrations in 10 eV declustering potential are presented in Fig. 1. Fig. 1(a) shows the spectrum with Cu2+(0.2 mM) and cytidine (0.4 mM), and Fig. 1(b) with Cu2+ (0.4 mM) and cytidine (0.8 mM). It is quite clear there is a big difference of the peak distribution. Fig. 1(a) shows the peaks at m/z 169.1, 185.1, corresponding to [CuL(MeOH)1-2]2+; 112.1, 244.4 and 487.5, assigned as [B+H]+ (B = cytosine), [L+H]+ and [L2+H]+; 368.0, 611.5 and 854.2, corresponding to [Cu(NO3)L1-3]+; and 305.3, 548.5, 791.3 and 275.0 assigned as [Cu(L–H)L0-2]+ and [CuL2]2+, respectively. There are also some other peaks, like 65.0, 126.1, 133.1 and 416.5 corresponding to [(MeOH)2+H]+, [Cu(CH2O)(MeOH)]+, [R]+ and [Cu(L–H)B]+. Except for the peaks, 112.1([B+H]+), 243.9([L+H]+), 486.9([L2+H]+), 168.9 ([CuL(MeOH)]2+), 185.1([CuL(MeOH)2]2+) and 274.8([CuL2]2+) in Fig. 1(a), Fig. 1(b) also shows a series of Cu2+–cytidine peaks, 396.3, 518.0, 639.5, 761.0, 882.7, 1004.7 and 1126.4, corresponding to [CuL3-9]2+, respectively. The chemical composition of these ions are assigned and confirmed from isotopic patterns, especially for those that contain Cu. The two isotopes 63Cu and 65Cu have an abundance ratio 69:31. The observed [B+H]+ ion in Fig. 1 would be formed through the dissociation of an N–glycoside bond of [L+H]+ at the interface of the instrument according to reaction (1).| | | [L+H]+ → [B+H]+ + [R–H] | (1) |
It is quite clear to see from Fig. 1 that the concentration will affect the distribution of the complexes of CuII–cytidine, and a higher concentration would cause more doubly charged cations to be produced. The reason could be relative with the complex stability constants in solution, and the higher concentration could be helpful to the predominant formation of the CuII–cytidine complexes. Moriwaki33 had investigated the complexes of CdII–guanosine by ESI-MS; he observed the molar ratio of Cd2+ to guanosine also had a dramatic effect on the ion peak distribution. Kearns et al. observed that the molar ratio and pH value affect the formation of the complexes in liquid in the magnetic resonance studies of Cu2+ interaction with nucleosides and nucleotides.64
From the ion peak distribution of [CuLn]2+ in Fig. 1(b), we can see that the peaks of n = 2 and 4 are the highest which means they both have a very stable coordination structure. This stable coordination structure would be relative with the characteristics of both cytidine and Cu2+. The N(3) and O(7) of cytidine are the preferred sites for metal to bind, and have been proved in many experiments.7,43,45 Binding to either N(3) or O(7) depends on the softness/hardness of the metal ions and possibly steric aspects. Sometimes the cytidine can bind metal simultaneously through N(3) and O(7) in chelating (or semichelating) fashion or bridge fashion.7 Generally, the CuII has a coordination number of 4, and prefers the square-planar or octahedral geometry while coordinating with some ligands.65–68 According to these facts and the calculation results, for the structure of [CuL2]2+, CuII coordinates with N(3) and O(7) simultaneously to form a planar structure with cytidine base; for [CuL4]2+, CuII only coordinates with N(3) to form a planar structure with cytidine base; for [CuL3]2+, CuII binds with 3 N(3) atoms in the same plane. For other complex cations, [CuLn]2+, we suggest that the extra ligands will loosely bind together through electrostatic strength based on structures of [CuL4]2+. The IR spectrophotometry results indicated that the metal cations bind N(3) and O(7) simultaneously with the cytidine in the solid chelates for the complexes [ML2]n+ (M = U(VI), Th(IV), Ce(III) and La(III)).69
3.2
CID of Cu2+-cytidine complex cations [Cu(L–H)]+ and [Cu(L–H)L]+
The cations of [Cu(L–H)]+ and [Cu(L–H)L]+ were observed in the ESI spectra of low concentration solution and 10 eV DP (Fig. 1(a)) .
The CID spectrum of [Cu(L–H)]+ at a collision energy of 14 eV is shown in Fig. 2. It shows that there is only one dissociation pathway for losing CH2O (m/z 275); and followed by elimination of H2O (m/z 257) or breaking the ribose ring to lose C2H3O2 (m/z 216) or CuC2H2O2 (m/z 154); then the higher order dissociation will continue and form other small daughter ions like m/z = 229, 173 and 174, etc. These dissociation processes were very similar to the chemical ionization mass spectrometry of nucleosides results.70–74 For the structure of [Cu(L–H)]+ (Table 1), the calculation results show that the coordination sites for copper(II) with cytidine are N3 and O(7), and the deprotonation site is an amino group on the base and not on the hydroxy group of the sugar moiety which is also consistent with previous results.34,45
Table 1 B3LYP/LANDL2DZ6-311+G(d) calculations of potential structures for the compounds of CuII–cytidine complexes
| Compound |
Structure |
| [Cu(L–H)]+ |
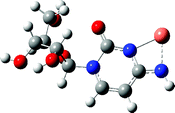
|
| [CuL(L–H)]+ |
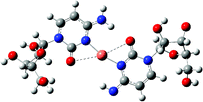
|
|
[CuL2]2+ |
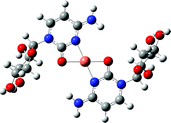
|
|
[CuL3]2+ |
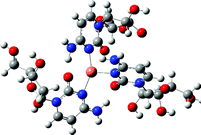
|
|
[CuL4]2+ |
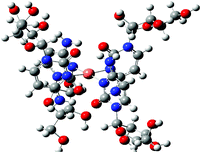
|
|
[CuL(MeOH)]2+ |
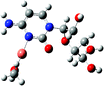
|
|
CID of the [CuL(L–H)]+ complex shows quite complicated fragmentation pathways, and there is no L or (L–H) loss channel . Fig. 3 is the CID spectrum of [CuL(L–H)]+ at a collision energy of 15 eV. Three main fragmentation channels were observed and identified as R–H (R = ribose) loss (m/z 416), reaction (2a); breaking the ribose ring to lose R–CH2O (m/z 445), reaction (2b) and to lose R–C2H4O (m/z 459), reaction (2c). Besides these, a minor channel of CH2O lost (m/z 518) was also observed, reaction (2d).
| | | [CuL(L–H)]+ → [Cu(L–H)(B+H)]+ + [R–H] | (2a) |
| | | → [CuL(B+COH)]+ + [R–CH2O] | (2b) |
| | | → [CuL(B+CH2COH)]+ + [R–C2H4O] | (2c) |
| | | → [CuL(L–H) –CH2O]+ + [CH2O] | (2d) |
![CID
spectra of [CuL(L–H)]+ at Elab = 18 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f3.gif) |
| | Fig. 3
CID
spectra of [CuL(L–H)]+ at Elab = 18 eV. | |
The calculation results show that the structure of [CuL(L–H)]+ (Table 1) is the one where the CuII coordinates with N(3) and O(7) simultaneously forming a planar structure with the cytidine base. This structure has high stability and the fragmentation only involves the ribose being broken.
The results of CID [CuL2-6]2+ complexes show that their fragmentation channels are quite different.
Fig. 4 shows the MS/MS spectra of [CuL2]2+ at a collision energy of 20 eV. Three main primary fragmentation channels were identified as, inter-ligand proton transfer accompanied by dissociation to give [L+H]+ and [Cu(L–H)]+, reaction (3a); [R–H] is lost to produce [CuLB]2+, reaction (3b); and dissociation charge reduction to break the N–glycoside bond of one ligand to give [CuL(B–H)]+ and [R]+, reaction (3c). Besides these, there is also a minor dissociation channel of dissociation charge reduction (DCR) to break the N–glycoside bond of one ligand to give [CuL(R–2H)]+ and [B+H]+, reaction (3d).
| | | [CuL2]2+ → [Cu(L–H)]+ + [L+H]+ | (3a) |
| | | → [CuL(R–2H)]+ + [B+H]+ | (3d) |
The calculation results show that the structure of [CuL2]2+ (Table 1) is where CuII coordinates with N(3) and O(7) simultaneously to form a planar structure. The fragmentations mainly involve the ribose being broken. During the fragmentation process, the inter-ligand electron transfer dissociation channel wasn't observed, even though Cu has the highest IE2 (20.3 ev)75 among all the transition metals, and the electron transfer from a neutral cytidine ligand (IP = 8.6 eV)70 to a double charge Cu2+ is energetically favorable. Cytidine is a protic solvent ligand with a quite high proton affinity (PA = 234.8 kcal mol−1)71 and may be the possible reason for inter-ligand proton transfer rather than electron transfer to occur.
Fig. 5 is the CID spectrum of [CuL3]2+ at collision energy of 16 eV. The CID process is quite different from [CuL2]2+, the inter-ligand proton transfer dissociation channel was mainly observed (reaction 4).
| | | [CuL3]2+ → [CuL(L–H)]+ + [L+H]+ | (4) |
Generally the complex stabilization could be achieved by multiple ligations which facilitates delocalization of the Cu2+ charges onto the cytidine ligands. However, an additional ligand also increases the possibility of other competing dissociation pathways, such as proton transfer, neutral ligand loss, etc. In this experiment, one more ligand adding from [CuL2]2+ to form [CuL3]2+ makes the inter-ligand proton transfer dissociation dominate. The reason is probably relative to the structure of [CuL3]2+. As we mentioned before, the molecule [CuL2]2+ is very stable for its high peak intensity in the ESI spectrum and possible stable four coordinated molecular structure. The addition of one more cytidine ligand changes the planar coordination structure (Table 1), and the CuII only binds with 3 N(3) atoms. This coordination structure is not very stable, and the very low energy collision makes it go back to the planar structure again to form [CuL(L–H)]+ and [L+H]+ rather than [CuL2]2+ and [L]. The reason is that the inter-ligand proton transfer is more favorable than the electron transfer, and the formation of [CuL(L–H)]+ and [L+H]+ is easier than the formation of [CuL2]2+ and [L].
The CID spectrum of [CuL4]2+ (m/z 518.0) at a collision energy of 24 eV is shown in Fig. 6. Only two primary dissociation channels could be identified, the inter-ligand proton transfer process forming [CuL(L–H)]+ and [L2+H]+ (reaction 5a) and [CuL2(L–H)]+ and [L+H]+ (reaction 5b). The structure for [CuL4]2+ (Table 1) is a CuII coordinating with four N(3) in the same plane, and the two CID channels are reasonable and the fragmentation products are quite stable. The [L+H]+ ions can also come from the dissociation of [L2+H]+ (reaction (6)) which was identified by the CID of [L2+H]+.
| | | [CuL4]2+→ [CuL(L–H)]+ + [L2 + H]+ | (5a) |
| | | → [CuL2(L–H)]+ + [L + H]+ | (5b) |
| | | [L2 + H]+ → [L+H]+ + [L] | (6) |
About the structure of [L2 +H]+, we assume that the two ligands bind together by hydrogen bonds. The structure of cytidine makes it possible to bind other ligands through hydrogen bonding. The hydrogen bonding guanine–cytidine or guanosine–cytidine pairs have been studied by several groups.57,72–74,76–78 Sindona et al.70 used the formation of proton-bound heterodimer between nucleosidesetc. to evaluate the proton affinities. A calculated lowest energy minimized structure of N–1-methylcytosine proton dimmer was given. The structure shows the occurrence of three hydrogen bonds between the O(7), N(3) and N(8). The proton-bound cytidine dimer observed in our experiment has a similar structure (see the supporting information†).
Fig. 7 shows the CID spectrum of [CuL5]2+ at a collision energy of 20 eV. The CID channels are different from other double charge cations, like [CuL2-4]2+. One more ligand adding from [CuL4]2+ to [CuL5]2+ makes the neutral ligand loss channel dominate, reaction (7a). Besides this, a small dissociation channel to produce a protonated cytidine dimer, reaction (7b), was also observed.
| | | [CuL5]2+ → [CuL4]2+ + [L] | (7a) |
| | | → [CuL2(L–H)]+ + [L2+H]+ | (7b) |
Fig. 8 is the CID spectrum of [CuL6]2+ at a laboratory energy of 16 eV. Three dissociation channels were identified. They are neutral ligand loss (reaction (8a)), inter-ligand proton transfer to produce protonated cytidine dimer [L2+H]+ and [CuL3(L–H)]2+ (reaction (8b)) and protonated cytidine trimer [L3+H]+ and [CuL2(L–H)]2+ (reaction (8c)).
| | | [CuL6]2+ → [CuL5]2+ + [L] | (8a) |
| | | → [CuL3(L–H)]+ + [L2 +H]+ | (8b) |
| | | → [CuL2(L–H)]+ + [L3 +H]+ | (8c) |
From the CID of the Cu2+–cytidine complex cations [CuL2-6]2+, we could see that the CuII mainly shows the coordination number 4 and has a tendency to coordinate with cytidine at N(3) or N(3) and O(7) to form a planar structure, and good examples are [CuL2]2+, [CuL(L–H)]+, [CuL4]2+ and [CuL3(L–H)]+; sometimes the CuII also shows the coordination number 3, and the typical example is [CuL3]2+, but this structure is not very stable and it is easy to lose a protonated L. For the structure of [CuLn]2+ (n > 4), we believe that the extra ligands will connect together with electrostatics and hydrogen bonds.
3.4
CID of Cu2+–cytidine-methane complex cations [CuL(MeOH)1-2]2+
The methanol molecule was also observed to be involved in the complex formation. [CuL(MeOH)1-2]2+ was observed in the ESI-MS spectrum of mixture of Cu2+ and cytidine (Fig. 1(a) and 1(b)). The CID spectra of [CuL(MeOH)]2+ (m/z 169.1) at a collision energy of 20 eV is shown in Fig. 9. Four main primary CID channels are observed: neutral methanol loss to from [CuL]2+ (m/z 153.1), reaction (9a), dissociation charge reduction to form [CuB(MeOH)]+ (m/z 205.1) and [R]+ (m/z 133.1), reaction (9b), dissociation charge reduction to produce [Cu(CH2OH)(MeOH)]+ (m/z 125.1) and [R–CH2OH]+ (m/z 213.1), reaction (9c), and dissociation charge reduction forming [Cu(MeOH)(BCOH)]+ (m/z 234.1), [C3H5O2]+ (m/z 73) and neutral radical CH2OH, reaction (9d). For [CuL(MeOH)2]2+, only MeOH loss is observed in its CID spectrum.| | | [CuLMeOH)]2+ → [CuL]2+ + [MeOH] | (9a) |
| | | → [CuB(MeOH)]+ + [R]+ | (9b) |
| | | → [Cu(CH2OH)(MeOH)]+ + [L–CH2OH]+ | (9c) |
| | | → [Cu(MeOH)(BCOH)]+ + [C3H5O2]+ + [CH2OH] | (9d) |
![CID
spectra of [CuL(CH3OH)]2+ at Elab = 10 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f9.gif) |
| | Fig. 9
CID
spectra of [CuL(CH3OH)]2+ at Elab = 10 eV. | |
The structure for [CuL(MeOH)]2+ shows that CuII has a coordination number of 3, N(3), O(7) of the L ligand and O of the MeOH. The fragmentation of [CuL(MeOH)]2+ mainly involves an extra MeOH loss or braking of the ribose. Reaction (9c) shows that the ligand base can also be lost. The structure of [CuL(MeOH)2]2+ shows that the CuII has a coordination number of 4, 2 with O of the MeOH molecules and 2 with N(3), O(7) of the ligand.
3.5 Comparison of the formation and CID of Cu2+–cytidine with Cu2+–guanosine
The formation of CuII–guanosine and its CID chemistry have been previously investigated.46 A comparison of results reported here of Cu2+–cytidine complexes indicates that these two molecules exhibit both similarities and differences in their formation and CID chemistry. Generally speaking, they both can form double charge complexes, [CuLn]2+ and [CuL(MeOH)m]2+, by the electronspray of the methanol and water solution of Cu2+ with ligands, and multiple competing dissociation channels occurred for [CuL2]2+, but the dissociation channels for [CuLn]2+ (n > 2) are quite simply dominated by one channel. The big difference of the CID chemistry for these two complexes is that the inter-electron transfer is the dominant channel for Cu2+–guanosine complexes, [CuLn]2+(n > 2), while the inter-ligand proton transfer dominates the CID process of Cu2+–cytidine complexes, [CuLn]2+(n > 2). For these big and complicated systems, it is too hard to provide the potential energy diagrams with energy barriers that bare no relationship to the final energies for the products through calculation, but the reasons could be attributed to their differences in ionization energy (IE) and proton affinity (PA). In other words, the balance between the attractive force of the hydrogen bonding and the repulsive electrostatic force determines the dissociation types.79 The IE for cytidine (IE = 8.6 eV) is 0.6 eV bigger than that of guanosine (IE = 8.0 eV), and the PAs for them are comparable PA (cytidine) = 234.8 kcal mol−1, PA (guanosine) = 236.8 kcal mol−1.77 The lower IE for guanosine makes the electron transfer preferable for CuII–guanosine complexes, [CuLn]2+ (n > 2), while the higher IE for cytidine makes the inter-proton transfer preferred for CuII–cytidine complexes, [CuLn]2+ (n > 2). The second difference is that, after coordination with Cu2+, guanosine still has enough sites to hydrogen-bond with other guanosine ligands, and there is no so-called magic number in CuII–guanosine complexes, [CuLn]2+. For cytidine, after coordination with CuII, there are no extra sites for hydrogen-bonding, and the extra ligands only bind together through weak electrostatics, so the magic number 2 and 4 exist for their stable complex configurations.
4. Conclusion
The singly charged cations, [L+H]+, [L2+H]+, and [B+H]+, and doubly charged cations, [CuLn]2+, [CuL(MeOH)n]2+, [Cu(L–H)Ln]+, (L = cytidine, B = cytosine) were formed by electronsprary of water and methanol solution of Cu(NO3)2 and cytidine. The collision-induced dissociations (CIDs) of [Cu(L–H)L0-1]+, [CuL2-6]2+ and [CuL(MeOH)1-2]2+ were investigated in low energy using the ESI-MS/MS instrument; and the structure, thermal energy and entropy for the most of these complexes were calculated with ab initio and DFT methods through Gaussian 09 program. The broken parts for the [Cu(L–H)L0-1]+ only on the ribose group of the ligand during the CID process, including ribose group loss ribose side chain and ribose ring braking. The inter-ligand proton transfer was observed in the CID of double charge CuII–cytidine complexes, [CuLn]2+ (n = 2–6), and other channels, including charge reduction dissociation and neutral ligand loss were also observed. The peak distribution for [CuL2-6]2+ shows that the Cu2+ likes to coordinate with N(3) or N(3) and O(7) to form a planar structure, which makes the [CuL2]2+ and [CuL4]2+ produce the highest peaks. Neutral MeOH loss and three kinds of charge reduction dissociation channels were presented in the CID of [CuL(MeOH)]2+. In the experiment, the Cu2+ shows the coordination number 3 or 4 in the complexes of CuII–cytidine and CuII–cytidine–MeOH. The N(3) and O(7) are mainly coordination sites for cytidine. The dimeric cytidine was produced in the CID of [CuLn]2+ (n = 4–6), and could be attributed to be the interactions of two cytidinesviahydrogen bonds. The inter-ligand electron transfer from ligand to Cu2+ to produce a ligand radical cation was not observed in the CID of the doubly charged CuII–cytidine complexes, [CuLn]2+ (n = 2–6), and the reasons can be attributed to the ligand's high ionization energy and proton affinity, which makes the inter-ligand proton transfer more favorable than electron transfer.
Acknowledgements
The authors gratefully acknowledge the financial support from General Foundation of Tianjin Science Committee for Applied Basic Research (No. 08JCYBJC00500) and the National Instrumentation Program (2011YQ170067).
References
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1205–1218 CrossRef CAS.
- S. S. Tan, S. J. Kim and E. T. Kool, J. Am. Chem. Soc., 2011, 133, 2664–2671 CrossRef CAS.
- P. Hirva, A. Nielsen, A. D. Bond and C. J. McKenzie, J. Phys. Chem. B, 2010, 114, 11942–11948 CrossRef CAS.
- J. C. Joyner and J. A. Cowan, J. Am. Chem. Soc., 2011, 133, 9912–9922 CrossRef CAS.
- S. J. Kim and E. T. Kool, J. Am. Chem. Soc., 2006, 128, 6164–6171 CrossRef CAS.
- A. Violante, V. Cozzolino, L. Perelomov, A. G. Caporale and M. Pigna, J. Soil. Sci. Plant. Nut., 2010, 10, 268–292 Search PubMed.
- L. Duchackova, D. Schroder and J. Roithova, Inorg. Chem., 2011, 50, 3153–3158 CrossRef CAS.
- A. Trabocchi, D. Scarpi and A. Guarna, Amino Acids, 2008, 34, 1–24 CrossRef CAS.
- B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487–516 CrossRef CAS.
- L. Lomozik, A. Gasowska, R. Bregier-Jarzebowska and R. Jastrzab, Coord. Chem. Rev., 2005, 249, 2335–2350 CrossRef CAS.
- H. Sigel, J. Inorg. Nucl. Chem., 1977, 39, 1903–1911 CrossRef CAS.
- H. Sigel, B. E. Fischer and B. Prijs, J. Am. Chem. Soc., 1977, 99, 4489–4496 CrossRef CAS.
- B. Knobloch and H. Sigel, JBIC, J. Biol. Inorg. Chem., 2004, 9, 365–373 CrossRef CAS.
- H. Sigel, C. P. Da Costa and R. B. Martin, Coord. Chem. Rev., 2001, 219–221, 435–461 CrossRef CAS.
- T. Theophanides and J. Bariyanga, J. Mol. Struct., 1989, 214, 177–184 CrossRef CAS.
- J. Swiatek-Kozlowska, J. Brasun, A. Dobosz, E. Sochacka and A. Glowacka, J. Inorg. Biochem., 2003, 93, 119–124 CrossRef CAS.
- Y. Kinjo, L. Ji, N. A. Corfu and H. Sigel, Inorg. Chem., 1992, 31, 5588–5596 CrossRef CAS.
- Y. Y. Chao and D. R. Kearns, J. Am. Chem. Soc., 1977, 99, 6425–6434 CrossRef CAS.
- A. T. Blades, P. Jayaweera, M. G. Ikonomou and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 1990, 102, 251–267 CrossRef CAS.
- A. T. Blades, P. Jayaweera, M. G. Ikonomou and P. Kebarle, J. Chem. Phys., 1990, 92, 5900–5906 CrossRef CAS.
- A. T. Blades, P. Jayaweera, M. G. Ikonomou and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 1990, 101, 325–336 CrossRef CAS.
- P. Jayaweera, A. T. Blades, M. G. Ikonomou and P. Kebarle, J. Am. Chem. Soc., 1990, 112, 2452–2454 CrossRef CAS.
- M. Peschke, A. T. Blades and P. Kebarle, J. Am. Chem. Soc., 2000, 122, 10440–10449 CrossRef CAS.
- S. E. Rodriguez-Cruz, R. A. Jockusch and E. R. Williams, J. Am. Chem. Soc., 1998, 120, 5842–5843 CrossRef CAS.
- J. A. Stone and D. Vukomanovic, Chem. Phys. Lett., 2001, 346, 419–422 CrossRef CAS.
- A. A. Shvartsburg and K. W. M. Siu, J. Am. Chem. Soc., 2001, 123, 10071–10075 CrossRef CAS.
- M. Peschke, A. T. Blades and P. Kebarle, J. Am. Chem. Soc., 2000, 122, 1492–1505 CrossRef CAS.
- Z. L. Cheng, K. W. M. Siu, R. Guevremont and S. S. Berman, Org. Mass Spectrom., 1992, 27, 1370–1376 CrossRef CAS.
- M. Kohler and J. A. Leary, J. Am. Soc. Mass Spectrom., 1997, 8, 1124–1133 CrossRef CAS.
- U. N. Andersen and G. Bojesen, Int. J. Mass Spectrom. Ion Processes, 1996, 153, 1–7 CrossRef CAS.
- B. J. Hall and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 1999, 10, 402–413 CrossRef CAS.
- A. A. Shvartsburg, J. G. Wilkes, J. O. Lay and K. W. M. Siu, Chem. Phys. Lett., 2001, 350, 216–224 CrossRef CAS.
- H. Moriwaki, J. Mass Spectrom., 2003, 38, 321–327 CrossRef CAS.
- S. Mochizuki and A. Wakisaka, J. Phys. Chem. B, 2003, 107, 5612–5616 CrossRef CAS.
- F. Moroni, A. Famulari, M. Raimondi and M. Sabat, J. Phys. Chem. B, 2003, 107, 4196–4202 CrossRef CAS.
- E. S. Baker, S. L. Bernstein and M. T. Bowers, J. Am. Soc. Mass Spectrom., 2005, 16, 989–997 CrossRef CAS.
- S. Guillaumont, J. Tortajada, J.-Y. Salpin and A. M. Lamsabhi, Int. J. Mass Spectrom., 2005, 243, 279–293 CrossRef CAS.
- M. Noguera, M. Sodupe and J. Bertran, Theor. Chem. Acc., 2004, 112, 318–326 CrossRef CAS.
- Tiziana Marino, Donatella Mazzuca, Nino Russo, Marirosa Toscano and A. Grand, Cryst. Growth Des., 2010, 110, 138–147 CAS.
- Y. Niu, S. Q. Feng, Rongjun Qu, Yunqiao Ding, Dengxu Wang and Yike Wang, Int. J. Quantum Chem., 2011, 111, 991–1001 CrossRef CAS.
- R. C. S. Dunbar, D. Jeffrey and Jos. Oomens, J. Am. Chem. Soc., 2011, 133, 9376–9386 CrossRef CAS.
- E. A. Motea and A. J. Berdis, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1151–1166 CrossRef CAS.
- B. Knobloch and H. Sigel, JBIC, J. Biol. Inorg. Chem., 2004, 9, 365–373 CrossRef CAS.
- B. Heddi and A. T. Phan, J. Am. Chem. Soc., 2011, 133, 9824–9833 CrossRef CAS.
- G. Bhaskar, M. A. Chary, M. K. Kumar, K. Syamasundar, M. Vairamani and S. Prabhakar, Rapid Commun. Mass Spectrom., 2005, 19, 1536–1544 CrossRef CAS.
- P. Cheng and D. K. Bohme, J. Phys. Chem. B, 2007, 111, 11075–11082 CrossRef CAS.
- W. A. Tao, D. Zhang, E. N. Nikolaev and R. G. Cooks, J. Am. Chem. Soc., 2000, 122, 10598–10609 CrossRef CAS.
- I. K. Chu, C. F. Rodriquez, T.-C. Lau, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. B, 2000, 104, 3393–3397 CrossRef CAS.
- I. K. Chu, C. F. Rodriguez, A. C. Hopkinson, K. W. Siu and T. C. Lau, J. Am. Soc. Mass Spectrom., 2001, 12, 1114–1119 CrossRef CAS.
- E. Bagheri-Majdi, Y. Ke, G. Orlova, I. K. Chu, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. B, 2004, 108, 11170–11181 CrossRef CAS.
- I. K. Chu, S. O. Siu, C. N. W. Lam, J. C. Y. Chan and C. F. Rodriquez, Rapid Commun. Mass Spectrom., 2004, 18, 1798–1802 CrossRef CAS.
- S. Wee, A. J. O'Hair Richard and W. D. McFadyen, Rapid Commun. Mass Spectrom., 2005, 19, 1797–1805 CrossRef CAS.
- Z. A. Tehrani, A. Fattahi and A. Pourjavadi, Carbohydr. Res., 2009, 344, 771–778 CrossRef CAS.
- Z. A. Tehrani, A. Fattahi and A. Pourjavadi, THEOCHEM, 2009, 913, 117–125 CrossRef CAS.
- W. Z. Shen and B. Lippert, J. Inorg. Biochem., 2008, 102, 1134–1140 CrossRef CAS.
- D. Buist, N. J. Williams, J. H. Reibenspies and R. D. Hancock, Inorg. Chem., 2010, 49, 5033–5039 CrossRef CAS.
- J. L. Sessler, J. Jayawickramarajah, M. Sathiosatham, C. L. Sherman and J. S. Brodbelt, Org. Lett., 2003, 5, 2627–2630 CrossRef CAS.
- R. Drouin, H. Rodriguez, S. W. Gao, Z. Gebreyes, T. R. O'Connor, G. P. Holmquist and S. A. Akman, Free Radical Biol. Med., 1996, 21, 261–273 CrossRef CAS.
- B. Halliwell and O. I. Aruoma, FEBS Lett., 1991, 281, 9–19 CrossRef CAS.
-
G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, B. Mennucci, H. N. G. A. Petersson, M. Caricato, X. Li, H. P. Hratchian, J. B. A. F. Izmaylov, G. Zheng, J. L. Sonnenberg, M. Hada, K. T. M. Ehara, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, O. K. Y. Honda, H. Nakai, T. Vreven, J. A. Montgomery Jr., F. O. J. E. Peralta, M. Bearpark, J. J. Heyd, E. Brothers, V. N. S. K. N. Kudin, R. Kobayashi, J. Normand, A. R. K. Raghavachari, J. C. Burant, S. S. Iyengar, J. Tomasi, N. R. M. Cossi, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, C. A. V. Bakken, J. Jaramillo, R. Gomperts, R. E. Stratmann, A. J. A. O. Yazyev, R. Cammi, C. Pomelli, J. W. Ochterski, K. M. R. L. Martin, V. G. Zakrzewski, G. A. Voth, J. J. D. P. Salvador, S. Dapprich, A. D. Daniels, J. B. F. O. Farkas, J. V. Ortiz, J. Cioslowski and D. J. Fox, Inc., Wallingford CT, 2009., GAUSSIAN 09 (Revision A.02), Gaussian, Inc.,Wallingford, CT, 2009 Search PubMed.
- V. Danilov, T. Vanmourik and V. Poltev, Chem. Phys. Lett., 2006, 429, 255–260 CrossRef CAS.
- M. Alcolea Palafox and Jessica Talaya, J. Phys. Chem. B, 2010, 114, 15199–15211 CrossRef CAS.
-
D. Feller, https://bse.pnl.gov/bse/portal.
- W. Lesniak, W. R. Harris, J. Y. Kravitz, J. Schacht and V. L. Pecoraro, Inorg. Chem., 2003, 42, 1420–1429 CrossRef CAS.
- V. Shuvaev Konstantin, S. M. Abedin Tareque, A. McClary Corey, N. Dawe Louise, L. Collins Julie and L. K. Thompson, Dalton Trans., 2009, 2926–2939 Search PubMed.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Phys. Chem. A, 2009, 113, 9559–9567 CrossRef CAS.
- R. R. Wright, N. R. Walker, S. Firth and A. J. Stace, J. Phys. Chem. A, 2001, 105, 54–64 CrossRef CAS.
- R. Ferreiros-Martinez, D. Esteban-Gomez, C. Platas-Iglesias, A. de Blas and T. Rodriguez-Blas, Dalton Trans., 2008, 5754–5765 RSC.
- B. Knobloch, C. P. Da Costa, W. Linert and H. Sigel, Inorg. Chem. Commun., 2003, 6, 90–93 CrossRef CAS.
- D. Armentano, G. De Munno, L. Di Donna, G. Sindona, G. Giorgi and L. Salvini, J. Am. Soc. Mass Spectrom., 2004, 15, 268–279 CrossRef CAS.
- N. S. Rannulu and M. T. Rodgers, Phys. Chem. Chem. Phys., 2005, 7, 1014–1025 RSC.
- M. Noguera, M. Sodupe and J. Bertran, Theor. Chem. Acc., 2007, 118, 113–121 CrossRef CAS.
- M. Krishnan and J. C. Smith, J. Am. Chem. Soc., 2009, 131, 10083–10091 CrossRef CAS.
- R. Improta and V. Barone, Theor. Chem. Acc., 2008, 120, 491–497 CrossRef CAS.
- M. Bruschi, L. De Gioia, R. Mitric, V. Bonacic-Koutecky and P. Fantucci, Phys. Chem. Chem. Phys., 2008, 10, 4573–4583 RSC.
- M. Noguera, J. Bertran and M. Sodupe, J. Phys. Chem. A, 2004, 108, 333–341 CrossRef CAS.
- D. Li, S. P. Song and C. H. Fan, Acc. Chem. Res., 2010, 43, 631–641 CrossRef CAS.
- C. K. Barlow, S. Wee, W. D. McFadyen and R. A. J. O'Hair, Dalton Trans., 2004, 3199–3204 RSC.
- Gronert. Scott, J. Mass Spectrom., 1999, 34, 787–797 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 
![CID
spectra of [Cu(L–H)]+ at Elab = 14 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f2.gif)
![CID
spectra of [CuL2]2+ at Elab = 20 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f4.gif)
![CID
spectra of [CuL3]2+ at Elab = 16 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f5.gif)
![CID
spectra of [CuL4]2+ at Elab = 24 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f6.gif)
![CID
spectra of [CuL5]2+ at Elab = 20 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f7.gif)
![CID
spectra of [CuL6]2+ at Elab = 10 eV.](/image/article/2012/RA/c2ra01293f/c2ra01293f-f8.gif)