Immobilization of chiral catalysts on magnetite nanoparticles for highly enantioselective asymmetric hydrogenation of aromatic ketones
Aiguo
Hu
,
Sophie
Liu
and
Wenbin
Lin
*
Department of Chemistry, CB#3290, University of North Carolina, Chapel Hill, North Carolina 27599, USA. E-mail: wlin@unc.edu
First published on 8th February 2012
Abstract
Magnetite nanoparticle (MNP)-supported asymmetric catalysts were synthesized by direct immobilization of a 4,4′-bisphosphonic acid-substituted BINAP-Ru-DPEN complex onto MNPs. The resulting heterogeneous catalyst (7) was highly active for the asymmetric hydrogenation of a wide range of aromatic ketones with remarkably high activity (>99% conversion with 0.1 mol% catalyst) and enantioselectivity (up to 98.1% ee) as a result of the bulky 4,4′-substituents (i.e., phosphonic acid groups) on BINAP. Catalyst 7 could be readily recycled and reused through simple magnetic decantation 9 times without the loss of activity or enantioselectivity. An alternative strategy was also explored to immobilize the [4,4′-(TMS)2-BINAP]-Ru-DPEN complex onto MNPs by first grafting a DPEN derivative onto the MNP surface followed by complexation of [4,4′-(TMS)2-BINAP]-Ru(DMF)2Cl2 to the surface-bound DPEN, but this immobilization strategy did not provide a robust heterogeneous catalyst, presumably due to oxidation of the Ru(II) intermediate by iron oxide nanoparticles.
Introduction
Asymmetric catalysis is one of the most powerful methods of producing enantioenriched compounds for academic research and industrial applications, particularly with respect to the biological processes, which are typically chirality-dependent.1–3 Numerous asymmetric catalysts have been designed along this line in the past four decades;1,2,4–10 however, their applications in industrial processes have often been hindered due to the high costs of both the precious metals and chiral ligands typical of these catalysts as well as difficulties in removing trace amounts of toxic metals from the organic products. Consequently, many heterogenization approaches have been explored to overcome these problems,7–11 including immobilization or incorporation of chiral catalysts onto organic polymers,11–20dendrimers,13inorganic oxide supports,11,21–25 metal–organic frameworks (MOFs),11,26–30 and biphasic systems or using special solvents such as ionic liquids and supercritical CO2.13,15,31–35 However, the heterogenized catalysts obtained from these methods are often less effective in terms of both activity and enantioselectivity than their homogeneous counterparts. There is thus a need to develop more efficient immobilization strategies for the heterogenization of homogeneous asymmetric catalysts.Previously, we synthesized a series of porous chiral catalystsvia a molecular building block approach for highly active heterogeneous asymmetric hydrogenation of aromatic ketones and keto esters36,37 and for enantioselective diethylzinc addition to aromatic aldehydes.38 However, significant fractions of the catalysts in these systems are buried inside the solids and are inaccessible to the organic substrates. It is well known that smaller particles have larger proportions of surface area. Because the surface area of a particle is inversely related to its diameter, immobilization of chiral catalysts onto smaller particles could result in a higher reactivity. For example, spherical nanoparticles 10 nm in diameter have a calculated surface area of 600 m2 cm−3, comparable to that of many of the porous materials used as supports for catalyst immobilization; consequently, they are expected to be just as effective. However, because of their small size, these nanoparticles are not easily separated from the reaction mixture by conventional means.39,40
The efficient and facile separation of magnetic particles from suspension by introducing an external magnetic field has been used to address this problem. In the last decade, the properties of magnetic nanoparticles have been investigated for use in protein binding and purification,41–43 MRI contrast enhancement,44–48drug targeting and delivery,46–48 and optical or photosensing,49 and in recent years, magnetic nanoparticles have been used in the immobilization of catalysts for a wide range of organic transformations.50–61 Previously, we successfully utilized an orthogonal functional group approach to immobilize a chiral catalyst, [Ru(4-P(O)(OH)2-(R)-BINAP)(DPEN)Cl2] (1) (DPEN is (R,R)-1,2-diphenylethylenediamine, Fig. 1), onto magnetite nanoparticles for asymmetric hydrogenation of aromatic ketones.50 The resulting nanocatalyst could be well dispersed in many solvents, and after catalysis, it could be easily recovered from the reaction mixture viamagnetic separation and reused up to 14 times. We believed that the chiral catalysts were attached to surface of the nanoparticlesvia4-phosphonic acid, while the orthogonal secondary chiral functionalities were then accessible for asymmetric catalysis. It was evident that the reactivity and enantioselectivity of these nanocatalysts (1-MNP) were comparable to those of the parent BINAP-Ru-DPEN complex 1.
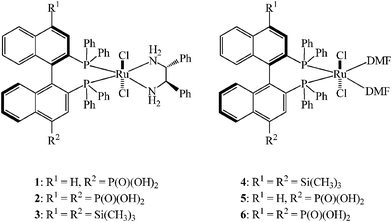 | ||
| Fig. 1 Chemical structures of Ru complexes used in this study. | ||
During our work on homogeneous asymmetric hydrogenation of aromatic ketones and aryl keto esters, we found that by introducing two bulky groups onto the 4,4′-positions of BINAP, the catalyst's enantioselectivity could be greatly enhanced; we termed this phenomenon as the “4,4′-effect” of BINAP derivatives.32,62–64 Because of the absence of the second bulky group on the BINAP, MNP-supported catalyst 1 did not exhibit as high enantioselectivities as the homogeneous 4,4′-bisphosphonic acid-substituted BINAP-Ru-DPEN catalyst 2 (Fig. 1). We hypothesized that introducing substituents in both positions would improve upon the enantioselectivity exhibited by 1-MNP. Herein, we wish to report our results on the highly active and enantioselective asymmetric hydrogenation of aromatic ketones catalyzed by 4,4′-substituted BINAP-Ru-DPEN complexes immobilized on magnetite nanoparticles.
Results and discussion
The synthetic route to make chiral catalysts immobilized on magnetite nanoparticles begins with treatment of 4,4′-bisphosphonic acid-substituted BINAP with [Ru(benzene)Cl2]2 followed by (R,R)-DPEN to afford the 4,4′-bisphosphonic acid-substituted BINAP-Ru-DPEN compound 2. Magnetite nanoparticles (MNPs) prepared in a co-precipitation manner65 were mixed with 2 under argon and characterized as previously described;50 the immobilization was completed after ultrasonication for 1 h (Scheme 1). The nanoparticles (7) collected via magnetic decantation were directly used for asymmetric hydrogenation of aromatic ketones.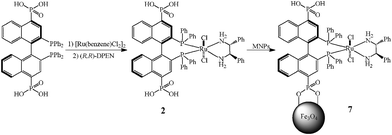
With the orthogonal secondary BINAP-Ru-DPEN moieties, these magnetite nanoparticles (7) exhibited exceptionally high reactivity and enantioselectivity in hydrogenation of aromatic ketones; 1-acetonaphthone was hydrogenated to α-(1-naphthyl)ethanol with complete conversion and 98.1% enantiomeric excess (ee) in isopropanol with 0.1 mol% loading of 7. A wide range of aromatic ketones were hydrogenated to their corresponding secondary alcohols with 7 with uniformly and remarkably high ee values of 91.4–98.1% (Table 1). This level of enantioselectivity is higher than that observed for both the parent homogeneous catalyst [Ru(BINAP)(DPEN)Cl2] and the MNP-immobilized monophosphonate catalyst 1-MNP (Table 1)50 and is comparable to the best results for this type of reaction among our work on 4,4′-BINAP family catalysts.32 The supernatant (before or after hydrogenation) was not reactive for hydrogenation of ketones (<0.01% conversion by GC), which indicated the heterogeneous nature of this reaction. The lack of Ru leaching into the supernatant (<2%) was also confirmed by inductively coupled plasma-mass spectrometric analysis. After diluting the highly viscous reaction mixture with equal amounts of either hexane or diethyl ether in order to facilitate their separation, these catalysts could be easily separated from reaction mixtures by introducing an external magnet, and pure product was obtained without any contamination of either ruthenium catalyst or MNPs.

| Substrate | 1 ee%b | 2 ee%b | 3 ee%b | 1-MNP ee%b | 7 ee% | 9 ee% | |
|---|---|---|---|---|---|---|---|
| Ar | R | ||||||
| a All of the reactions were carried out at rt with 0.1 mol% catalyst and 1% KOtBu under 700 psi hydrogen pressure at 25 °C in 20 h. The absolute configurations of the products are identical to those obtained by Ru-(R)-BINAP-(R,R)-DPEN. All the conversions were >99% as judged by the integrations of GC peaks. b These data are quoted from our previous results50,62 for comparison. | |||||||
| Ph | Me | 87.0 | 96.0 | 97.1 | 81.7 | 94.3 | 95.4 |
| 1-Naph | Me | 98.0 | 99.0 | 99.0 | 97.6 | 98.1 | 98.8 |
| 2-Naph | Me | 84.9 | 96.7 | 97.9 | 82.0 | 95.8 | 95.7 |
| 4-tBu-Ph | Me | 94.1 | 99.0 | 99.1 | 91.1 | 98.3 | 98.2 |
| 4-Me-Ph | Me | 85.6 | 95.4 | 97.4 | 80.5 | 95.7 | 93.6 |
| 4-Cl-Ph | Me | 75.9 | 90.5 | 95.8 | 70.6 | 91.4 | 90.0 |
| 4-MeO-Ph | Me | 82.5 | 95.8 | 95.4 | 77.7 | 93.8 | 90.0 |
| Ph | Et | 89.0 | 96.8 | 94.2 | 86.3 | 91.7 | 96.8 |
We have also successfully reused 7 systems for asymmetric hydrogenation of 1-acetonaphthone without the deterioration of enantioselectivity. As shown in Table 2, the 7 system was used for ten cycles of hydrogenation without any loss of enantioselectivity. The activity did not decrease for the first nine runs but began to drop at the tenth run. This loss of activity may not reflect the intrinsic instability of the magnetite-immobilized solid catalyst; the catalyst recycling and reuse experiments were conducted without rigorous exclusion of air, and the oxygen sensitivity of the ruthenium hydride complexes may have contributed to the loss of activity after multiple runs. Nonetheless, 1-acetonaphthone was hydrogenated with a total turnover number (TON) of ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and consistently high ee values.
000 and consistently high ee values.
| Run | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| ee% | 98.1 | 97.2 | 97.4 | 97.5 | 98.0 | 98.0 | 98.0 | 97.9 | 98.0 | 97.1 |
| conv.% | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 85 |
As shown in Fig. 2, after 2 is bound to the MNP, the second phosphonic acid group is free to bind to other MNP surface binding sites, which can result in severe aggregation of nanoparticles. Transmission electron microscopy (TEM) images showed MNPs formed heavy aggregation after the incorporation of Ru complex 2. We also found that the reactivity of this nanocatalyst decreased substantially if the catalyst was vacuum-dried or left in a solvent for a few weeks. We believe that the irreversible aggregation of the nanocatalysts formed through this process greatly decreased the number of surface groups available for catalysis. This dual-side binding ability of 2 to magnetite nanoparticles thus limited its long-term storage and presented a problem for its practical applications.
 | ||
| Fig. 2 TEM image of 7 showing its tendency to aggregate upon drying. | ||
Based on the structural features of the BINAP-Ru-DPEN complex, we reasoned that this problem could be solved by replacing 4,4′-bisphosphonic acid groups on BINAP with two inert substituents while installing one phosphonic acid moiety on the DPEN side. According to our studies on homogeneous asymmetric hydrogenation of ketones, 4,4′-(TMS)2-BINAP is the best choice.32
The DPEN-coated MNPs were synthesized following a strategy similar to that of a literature procedure for grafting DPEN onto silica materials.6610-Bromodecylphosphonic acid was mixed with MNPs in methanol, and the immobilization was completed after ultrasonication for 2 h. The surface-grafted MNPs were then heated with DPEN in anhydrous ethanol overnight to afford DPEN-coated MNPs (8). Under an argon atmosphere, freshly prepared [Ru(4,4′-(TMS)2-BINAP)(DMF)2Cl2] (4)31 was then stirred with a suspension of 8 in DMF at elevated temperature for 3 h to give the final superparamagnetic nanocatalyst (9) (Scheme 2).
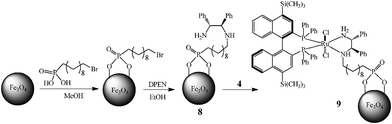 | ||
| Scheme 2 Immobilization of chiral Ru catalyst 4 on MNPsvia the DPEN linkage. | ||
The hydrogenations of aromatic ketones catalyzed by 9 were conducted under identical conditions. With 0.1 mol% nanocatalyst loading, all the aromatic ketones we tested were reduced to their corresponding secondary alcohols with complete conversion and 90.0–98.8% ee, which are comparable to those of the homogeneous catalyst [Ru(4,4′-(TMS)2-BINAP)(DPEN)Cl2] (3). The catalyst reuse experiments, however, were disappointing; only 60% conversion of 1-acetonaphthone was achieved with 98.3% ee for the second use of catalyst, and for the third use, there was no conversion at all. We believe that only a small fraction of the Ru complex was actively catalysing the asymmetric hydrogenation of aromatic ketones and was quickly deactivated during the hydrogenation reactions and the workup of the reactions. It is known that MNPs are comprised of iron(III) and iron(II) and that iron(III) is a strong oxidant with an Fe3+ to Fe2+ reduction potential of E° = 0.771 V. Because [Ru(BINAP)(DMF)2Cl2] is much less tolerant of oxidants than the corresponding BINAP-Ru-DPEN complex [Ru(BINAP)(DPEN)Cl2], much of 4 could be oxidized by iron(III) on the surface of the MNPs before the formation of the more stable DPEN complex with surface-bound DPEN. This kind of phenomenon took place when we attempted to immobilize 5 and 6 onto MNPs for asymmetric hydrogenation of β-keto esters. No conversion was obtained from either nanocatalyst. Control experiments showed that after treatment with MNPs for 2 h, 4 had no catalytic activity with respect to hydrogenation of methyl acetoacetate; however, untreated 4 could catalyze the hydrogenation to give complete conversion and 98.4% ee under identical conditions. This experiment unambiguously showed the instability of 4 (and its analogues 5 and 6) to MNPs. Non-oxidative spherical coated magnetic nanoparticles such as magnetite-embedded silica nanoparticles67 could be used to address this instability problem.
Conclusions
We have designed and synthesized MNP-supported chiral catalysts containing 4,4′-bisphosphonic acid-substituted BINAP-Ru-DPEN moieties (7). Due to the presence of bulky phosphonic acid groups as 4,4′-substituents, these chiral nanoparticles heterogeneously catalyzed the asymmetric hydrogenation of aromatic ketones with practically useful and remarkably high activity and enantioselectivity (up to 98.1% ee), exceeding the enantioselectivity of the analogous monophosphonic acid-substituted catalyst. Catalyst 7 could be readily recycled and reused 9 times with comparable activity or enantioselectivity via simple magnetic decantation with the application of an external magnet. However, this catalyst tended to aggregate upon storage due to the ability of its second phosphonic acid substituent to bind to other MNPs. Therefore, an alternate approach to synthesizing MNP-supported 4,4′-substituted BINAP-Ru-DPEN-based catalysts was investigated by coating MNPs with a DPEN derivative, then complexing the surface-bound DPEN with [Ru(4,4′-(TMS)2-BINAP)(DMF)2Cl2]. However, this approach failed to produce a robust heterogeneous catalyst for asymmetric hydrogenation of aromatic ketones, presumably as a result of the susceptibility of the Ru(II) intermediate to oxidation by the MNPs. Future efforts are directed at immobilization of these highly active and enantioselective catalysts on silica-coated magnetite particles.Experimental section
Materials and general procedures
All of the chemicals were obtained from commercial sources and used without further purification. All of the reactions and manipulations were carried out under argon with the use of standard inert atmosphere and Schlenk techniques. Solvents used in all the reactions were dried by standard procedures.68NMR spectra were recorded on a Bruker NMR 400 DRX spectrometer. 1H NMR spectra were recorded at 400 MHz and referenced to the proton resonance resulting from incomplete deuteration of chloroform-d1 (δ 7.26). 13C{1H} NMR spectra were recorded at 100 MHz, and all of the chemical shifts are reported downfield in ppm relative to the carbon resonance of chloroform-d1 (δ 77.0). 31P{1H} NMR were recorded at 161 MHz, and all of the chemical shifts are reported in ppm relative to the phosphorus resonance of 85% phosphonic acid (external standard). TEM images were taken with a Phillips CM 12 electron microscope at 100 kV. Ligands 1 and 2 and complexes 3–6 were synthesized according to literature procedures.69MNPs were synthesized according to literature procedures65 and characterized as previously described50 and washed thoroughly with methanol and dried under vacuum before use. The conversions and ee values were determined by GC on a Supelco β-Dex 120 column, 30 m × 0.25 mm × 0.25 μm; the detailed characterization of all of the hydrogenation products were reported previously.50,62Preparation of MNP-immobilized catalyst 7
The Ru complex 2 (2.4 mg, 2.0 μmol) and MNPs (60 mg) in anhydrous methanol (1 mL) were ultrasonicated under argon for 1 h. The solid collected by magnetic decantation was washed with methanol 3 times and dried under vacuum. The supernatants were combined and used for NMR analysis. With PPh3 (1.0 mg, 4.0 μmol) as the internal standard, 31P NMR spectra indicated that there was less than 5% of BINAP derivatives in the supernatants. The surface coverage of Ru catalyst on MNPs was thus estimated to be ∼33 μmol g−1.General procedure for asymmetric hydrogenation of ketones with MNP-immobilized catalysts
The immobilized catalyst (2 μmol) and potassium tert-butoxide (2.3 mg, 20 μmol) were weighed into a Teflon septum-sealed vial (reaction flask) in an inert atmosphere drybox, and then 1-acetonaphthone (0.34 mL, 2.0 mmol) and anhydrous isopropanol (1 mL) were added under argon. The vial was quickly transferred into a stainless steel autoclave and sealed. After purging with hydrogen 6 times, the final hydrogen pressure was adjusted to 700 psi. After 20 h, hydrogen pressure was released, and water (10 mL) and diethyl ether (2 mL) were added. The organic layer was separated out and passed through a mini silica gel column. An aliquot was analyzed on GC to determine conversion and ee. The absolute configurations of enantioenriched products from the present experiments were assigned on GC to be same as those samples obtained from (R, RR)-BINAP-Ru-DPEN-catalyzed reactions.General procedure for catalyst reuse experiments
Anhydrous degassed diethyl ether (1 mL) was added to the reaction mixture after hydrogenation in order to decrease the viscosity of the reaction mixture, thereby facilitating the subsequent separation of the catalyst from the reaction mixture. The catalyst was collected either via magnetic decantation or centrifugation, and the liquid layer was siphoned out. The residual solid was washed with anhydrous degassed isopropanol:diethyl ether (1:1) twice and then dried under vacuum. The ketone substrate, potassium tert-butoxide and isopropanol were then charged for another round of the hydrogenation reaction.
Preparation of MNP-immobilized catalyst 9
MNPs (80 mg) and 10-bromodecylphosphonic acid (30 mg, 0.1 mmol) were mixed with ethanol (5 mL). The resulting suspension was ultrasonicated for 2 h and then centrifuged. The solid collected by magnetic decantation was washed with ethanol 3 times and dried under vacuum. The supernatants were combined and used for NMR analysis. With PPh3 (2.7 mg, 0.010 mmol) as the internal standard, 31P NMR spectra indicated that there was 0.06 mmol of 10-bromodecylphosphonic acid in the supernatants. The surface coverage of 4 on magnetite nanoparticles was thus estimated to be ∼0.04 mmol. To the coated MNPs, (R,R)-DPEN (8.4 mg, 0.040 mmol), triethylamine (0.1 mL) and anhydrous methanol (5 mL) were added. This mixture was refluxed under argon overnight, and then the supernatant was removed to give DPEN-attached MNPs (8). Under argon, 8 was mixed with freshly prepared [Ru(4,4′-(TMS)2-BINAP)(DMF)2Cl2] (4) solution31 in DMF under argon and stirred at 80 °C for 3 h. The solid collected by magnetic decantation was washed with anhydrous methanol 3 times and dried in vacuo. The supernatants were combined and oxidized with hydroperoxide (30% aqueous solution) for 1 h and then used for NMR analysis. With PPh3 (5.3 mg, 0.020 mmol) as the internal standard, 31P NMR spectra indicated that there was 0.035 mmol of BINAP derivatives in the supernatants. The surface coverage of the Ru complex on the MNPs was estimated to be ∼5 μmol.General procedure for control experiments on stability of [Ru(BINAP)(DMF)2Cl2]-derived complexes
Freshly prepared [Ru(4,4′-(TMS)2-BINAP)(DMF)2Cl2] (4) (0.02 mmol) was dissolved in anhydrous degassed methanol (6 mL) and then split into 3 vacuum-dried Schlenk flasks (labeled A, B and C). To flasks B and C, freshly prepared, vacuum-dried MNPs (80 mg) were added under argon and then stirred under argon for 2 h. Flask C was sealed and centrifuged for 15 min, and the supernatant was carefully transferred to flask D with use of a cannula under argon. To flasks A, B and D, methyl acetoacetate (0.14 mL, 1.3 mmol, ca. 200 substrate/catalyst loading) was added. These three flasks were quickly transferred to a stainless steel Parr® reactor, and hydrogenation was performed under 100 atm hydrogen pressure. After 20 h, hydrogen was carefully released, and methanol was removed under reduced pressure. The residue was loaded on a mini silica gel column and washed with diethyl ether. The conversions of starting material to product were determined through NMR analysis, while the ee% values (if available) were obtained through chiral GC analysis. A: 100% conversion, 98.4% ee; B: 0% conversion; D: 0% conversion. This experiment clearly showed the instability of complex 8 to magnetite nanoparticles.Acknowledgements
We thank the National Science Foundation (CHE-1111490) for financial support. SL is supported by a UNC William W. and Ida W. Taylor Fellowship.References
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS and references therein.
- W. S. Knowles, Adv. Synth. Catal., 2003, 345, 3 CrossRef CAS.
- V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734 CrossRef CAS.
- Y.-G. Zhou, W. Tang, W.-B. Wang, W. Li and X. Zhang, J. Am. Chem. Soc., 2002, 124, 4952 CrossRef CAS.
- T. Ireland, K. Tappe, G. Grossheimann and P. Knochel, Chem.–Eur. J., 2008, 14, 3509 CrossRef CAS.
- N. W. Boaz, S. D. Debenham, E. B. Mackenzie and S. E. Large, Org. Lett., 2002, 4, 2421 CrossRef CAS.
- J. M. Thomas, T. Maschmeyer, B. F. G. Johnson and D. S. Shephard, J. Mol. Catal. A: Chem., 1999, 141, 139–144 CrossRef CAS.
- D. Sinou, Adv. Synth. Catal., 2002, 344, 221 CrossRef CAS.
- C. E. Song and S. G. Lee, Chem. Rev., 2002, 102, 3495 CrossRef CAS.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
- J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819 CrossRef CAS and references therein.
- Q. H. Fan, Y. M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS.
- L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS.
- C. Saluzzo and M. Lemaire, Adv. Synth. Catal., 2002, 344, 915 CrossRef CAS.
- L. Xu, K. H. Lam, J. Ji, J. Wu, Q. H. Fan, W. H. Lo and A. S. C. Chan, Chem. Commun., 2005, 1390 RSC.
- C. Dong, J. Zhang, W. Zheng, L. Zhang, Z. Yu, M. C. K. Choi and A. S. C. Chan, Tetrahedron: Asymmetry, 2000, 11, 2249 CrossRef.
- M. Nielsen, A. H. Thomsen, T. R. Jensen, H. J. Jakobsen, J. Skibsted and K. V. Gothelf, Eur. J. Org. Chem., 2005, 342 CrossRef CAS.
- W.-S. Huang, Q.-S. Hu and L. Pu, J. Org. Chem., 1999, 64, 7940 CrossRef CAS.
- H.-S. Yu, Q.-S. Hu and L. Pu, J. Am. Chem. Soc., 2000, 122, 6500 CrossRef CAS.
- S. Xiang, Y. L. Zhang, Q. Xin and C. Li, Chem. Commun., 2002, 2696 RSC.
- S. Huh, J. W. Wiench, B. G. Trewyn, S. Song, M. Pruski and V. S. Lin, Chem. Commun., 2003, 2364 RSC.
- D. J. Mihalcik and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 6229 CrossRef CAS.
- M. D. Jones, R. Raja, J. M. Thomas, B. F. G. Johnson, D. W. Lewis, J. Rouzaud and K. D. M. Harris, Angew. Chem., Int. Ed., 2003, 42, 4326 CrossRef CAS.
- S. J. Bae, S.-W. Kim, T. Hyeon and B. M. Kim, Chem. Commun., 2000, 31 CAS.
- C. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS.
- S. H. Cho, B. Q. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2565 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef CAS.
- H. L. Ngo, A. Hu and W. Lin, Chem. Commun., 2003, 1912 RSC.
- A. Hu, H. L. Ngo and W. Lin, Angew. Chem., Int. Ed., 2004, 43, 2501 CrossRef CAS.
- H. L. Ngo, A. Hu and W. Lin, Tetrahedron Lett., 2005, 46, 595–597 CrossRef CAS.
- L. Xu, and J. Xiao, in Asymmetric Catalysis in Ionic Liquids, ed. M. Benaglia, Recoverable and Recyclable Catalysts, Wiley, West Sussex, UK, 2009, pp. 259–300 Search PubMed.
- R. A. Brown, P. Pollet, E. McKoon, C. A. Eckert, C. L. Liotta and P. G. Jessop, J. Am. Chem. Soc., 2001, 123, 1254 CrossRef CAS.
- A. Hu, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2003, 125, 11490 CrossRef CAS.
- A. Hu, H. L. Ngo and W. Lin, Angew. Chem., Int. Ed., 2003, 42, 6000 CrossRef CAS.
- H. L. Ngo, A. Hu and W. Lin, J. Mol. Catal. A: Chem., 2004, 215, 177–186 CrossRef CAS.
- I. Gunjishima, T. Inoue and A. Okamoto, Langmuir, 2008, 24, 2407–2411 CrossRef CAS.
- J. M. Bell and F. K. Cameron, J. Phys. Chem., 1906, 10, 658–674 CrossRef.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938 CrossRef CAS.
- H. Gu, Z. Yang, J. Gao, C. K. Chang and B. Xu, J. Am. Chem. Soc., 2005, 127, 34 CrossRef CAS.
- J. S. Kim, C. A. Valencia, R. Liu and W. Lin, Bioconjugate Chem., 2007, 18, 333–341 CrossRef CAS.
- Y.-W. Jun, Y.-M. Huh, J.-S. Choi, J.-H. Lee, H.-T. Song, S.-J. Kim, S. Yoon, K.-S. Kim, J.-S. Shin, J.-S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732 CrossRef CAS.
- Y. Piao, A. Burns, J. Kim, U. Wiesner and T. Hyeon, Adv. Funct. Mater., 2008, 18, 3745–3758 CrossRef CAS.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552–557 CrossRef CAS.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS.
- N. Kohler, G. E. Fryxell and M. Zhang, J. Am. Chem. Soc., 2004, 126, 7206 CrossRef CAS.
- F. E. Osterloh, J. Am. Chem. Soc., 2002, 124, 6248 CrossRef CAS.
- A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486 CrossRef CAS.
- N. T. S. Phan and C. W. Jones, J. Mol. Catal. A: Chem., 2006, 253, 123–131 CrossRef CAS.
- C. S. Gill, B. A. Price and C. W. Jones, J. Catal., 2007, 251, 145–152 CrossRef CAS.
- M. Shokouhimehr, Y. Piao, J. Kim, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039–7043 CrossRef CAS.
- C. O. Dalaigh, S. A. Corr, Y. K. Gun'ko and S. J. Connon, Angew. Chem., Int. Ed., 2007, 46, 4329–4332 CrossRef CAS.
- O. Gleeson, R. Tekoriute, Y. K. Gun'ko and S. J. Connon, Chem.–Eur. J., 2009, 15, 5669–5673 CrossRef CAS.
- S. Shylesh, W. R. Thiel and V. Schünemann, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS.
- Y. Sun, G. Liu, H. Gu, T. Huang, Y. Zhang and H. Li, Chem. Commun., 2011, 47, 2583–2585 RSC.
- D. Bai, Q. Wang, Y. Song, B. Li and H. Jing, Catal. Commun., 2011, 12, 684–688 CrossRef CAS.
- N. T. S. Phan and H. V. Le, J. Mol. Catal. A: Chem., 2011, 334, 130 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihrit, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef CAS and references therein.
- K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13 CAS and references therein.
- A. Hu, H. L. Ngo and W. Lin, Org. Lett., 2004, 6, 2937 CrossRef CAS.
- A. Hu and W. Lin, Org. Lett., 2005, 7, 455 CrossRef CAS.
- M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801 CrossRef CAS.
- Y. Sahoo, H. Pizem, T. Fried, D. Golodnitsky, L. Burstein, C. N. Sukenik and G. Markovich, Langmuir, 2001, 17, 7907 CrossRef CAS.
- A. Ghosh and R. Kumar, J. Catal., 2004, 228, 386–396 CrossRef CAS.
- G. Liu, H. Gu, Y. Sun, J. Long, Y. Xu and H. Li, Adv. Synth. Catal., 2011, 353, 1317 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, 4th edn, Reed Educational and Professional Publishing Ltd, 1996 Search PubMed.
- M. Kant, S. Bischoff, R. Siefken, E. Gruendemann and A. Koeckritz, Eur. J. Org. Chem., 2001, 477 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |