Ionic radius-dependent self-assembly of closed/opened molecular capsules based on pentacyclopentanocucurbit[5]uril†
Ying-Feng
Hu
ab,
Kai
Chen
b,
Rui-Lian
Lin
a,
Wen-Qi
Sun
a,
Jing
Zhu
a,
Jing-Xin
Liu
*a,
Sai-Feng
Xue
b,
Qian-Jiang
Zhu
b and
Zhu
Tao
*b
aCollege of Chemistry and Chemical Engineering, Anhui University of Technology, Maanshan, 243002, China. E-mail: jxliu411@ahut.edu.cn (J.-X. Liu); Fax: 86-555-2311552
bKey Laboratory of Macrocyclic and Supramolecular Chemistry of Guizhou Province, Guizhou University, Guiyang 550025, P. R. China. E-mail: gzutao@263.net (Z. Tao); Fax: 86-851-3620906
First published on 18th April 2012
Abstract
Three coordination architectures, namely, {Ca2(H2O)4[(NO3)⊂Q*[5]]}2(NO3)6·26H2O (1), {Sr2(H2O)3(NO3)[(NO3)⊂Q*[5]]}(NO3)2·7H2O (2), {Ba(H2O)[(H2O)2 ⊂ Q*[5]]}–(NO3)2·11H2O (3), have been synthesized by reactions of the corresponding alkaline-earth metal salts with pentacyclopentanocucurbit[5]uril (Q*[5]). These compounds were characterized by single-crystal X-ray diffraction studies, showing that the ionic radius of the alkaline-earth metal plays a crucial role in determining which type of molecular capsule forms. Compound 1 is an incompletely closed molecular capsule, compound 2 corresponds to a closed molecular capsule, and compound 3 resembles an opened molecular capsule. The thermogravimetric analysis (TGA) of compound 2 was also characterized.
Introduction
The controlled self-assembly of precursor metal ions and organic ligands into coordination architectures has been a very active research area in the past decade.1,2 Interest in the area is justified by the useful electronic, magnetic, catalytic, or host–guest properties of the predictable coordination architectures.3–6 To date, the vast majority of reported constructions of coordination architectures focused on ligand design and synthesis. The main reason is that ligands are ideal tools for controlling geometry and spaces. In addition, ligand design provides new opportunities for introducing functionality. In contrast to the well-development on ligand design and synthesis, efforts on the selection of the metal ion serve as control factor in construction of coordination architectures are still significant.7 Ionic radius-dependent self-assembly remains an important research topic.As a type of macrocyclic host, cucurbit[n]urils8 (n = 5–8, 10, abbreviated as Q[n]) have attracted increasing interest. With a range of fairly rigid hydrophobic cavities, they display outstanding recognition towards hosting positively charged guest species, and the related host–guest chemistry has been extensively investigated and reviewed by pioneers.9 Furthermore, due to the presence of polarized carbonyl groups, Q[n] exhibits wonderful metal–ion recognition abilities. Q[n] can act as polydentate ligands and coordinate with many types of metal ions, such as alkali, alkaline-earth, and lanthanide metal ions.10–13 However, despite examples of the coordination architectures of Q[n] with metal ions, in neither case was the ionic radius-dependent self-assembly of Q[n] architectures observed. The influence of the ionic radius of the metal on the self-assembly process is still less well understood and systematic investigations are necessary.
Recently our research interests in this area has been focused on Q[n] derivatives because they usually exhibit better solubilities in aqueous media. In this paper, we present three coordination architectures of the pentacyclopentanocucurbit[5]uril14 ligand (here after abbreviated as Q*[5], Fig. 1), one derivative of Q[5] with three different alkaline-earth metal ions (Ca2+, Sr2+, Ba2+). The analysis of the crystallographic data of the three crystal structures reported clearly shows that the ionic radius of the alkaline-earth metal plays a crucial role in closing and opening the Q*[5]-based molecular capsule.
![Q*[5] and its single-crystal X-ray structure.](/image/article/2012/RA/c2ra20569f/c2ra20569f-f1.gif) | ||
| Fig. 1 Q*[5] and its single-crystal X-ray structure. | ||
Results and discussion
Description of the crystal structures
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Single-crystal X-ray diffraction analysis reveals that compound 1 is an incompletely closed molecular capsule structure. The perspective view of compound 1 is shown in Fig. 2.
space group. Single-crystal X-ray diffraction analysis reveals that compound 1 is an incompletely closed molecular capsule structure. The perspective view of compound 1 is shown in Fig. 2.
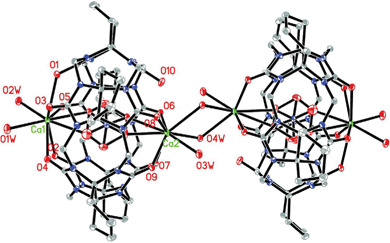 | ||
| Fig. 2 ORTEP diagram of compound 1; displacement ellipsoids are drawn at the 30% probability level. Solvate water molecules and nitrate anions are omitted for clarity. | ||
As can be seen in Fig. 2, there exists two crystallographically distinct Ca2+ ions in the asymmetric unit of compound 1. The Ca1 is coordinated by five carbonyl groups (O1, O2, O3, O4, and O5) at one of the portals of the Q*[5] ligand with Ca⋯O distances of 2.4458(14)–2.6319(14) Å, and two water molecules (O1w and O2w) from outside with a distance of 2.4103(17) and 2.3824(15) Å. The Ca2 leans toward the other side of the portal; being coordinated to only four carbonyl groups (O6, O7, O8 and O9) with Ca⋯O distances of 2.4163(15)–2.6764(16) Å. Three water molecules (O3w, O4w and O4wa) are also bound to the metal ion from outside with distances of 2.3727(17)–2.6023(16) Å. A disordered nitrate anion resides in the cavity of Q*[5] and is coordinated to the calcium cations (Ca1 and Ca2) from the inside. The most remarkable feature in compound 1 is that the Q*[5] molecule and the encapsulated nitrate anion, as well as two calcium cations form an incompletely closed molecular capsule {Ca2(H2O)4[(NO3)⊂Q*[5]]}3+. The special structure can be attributed to the fact that the calcium cation is not large enough to cover the portal of the Q*[5] molecule completely. Normally Q*[5] have two identical carbonyl-fringed portals of about 1.2 Å radius (take into account the van der Waals radii of the relevant atoms), which is slightly larger than that of the calcium cation (1.12 Å). In the case of compound 1, the Q*[5] molecule experiences a slight deformation, leading to one portal of the Q*[5] molecule being much larger than the other one. As a result, one portal of the Q*[5] molecule is closed by the calcium cation, whereas the other portal of the Q*[5] molecule cannot be sealed by the calcium cation completely.
It is interesting to remark that two neighboring incompletely closed molecular capsules are linked together through two bridging water molecules (O4w and O4wa), forming a dimer {Ca2(H2O)4[(NO3) ⊂ Q*[5]]}26+. In the crystal lattice of compound 1, each discrete dimer is surrounded by numerous nitrate anions and water molecules. These dimers are further connected through intermolecular hydrogen-bonds to form a complicated supramolecular network.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio yielded good quality single crystals of compound 2, which has the same space group as compound 1. Single-crystal X-ray diffraction analysis revealed that compound 2 is a coordination polymer constructed from closed molecular capsules. The asymmetric unit of compound 2 consists of two crystallographically distinct Sr2+ ions, a Q*[5] ligand, four nitrate anions, three coordinating water molecules and three uncoordinated water molecules. As shown in Fig. 3, Sr1 and Sr2 both have nine coordinate bonds. Each portal of the Q*[5] molecule is covered by one strontium cation, generating a completely closed molecular capsule {Sr2(H2O)3(NO3)[(NO3)⊂Q*[5]]}2+. The formation of the completely closed molecular capsule can ascribed to the size complementarity of the strontium cation and the Q*[5] portal. Interestingly, a nitrate anion is encapsulated in the cavity of the Q*[5] molecule. The distances between the nitrogen atom of the captured nitrate anion and the two strontium cations are 3.650 Å and 3.478 Å, respectively, indicating that the nitrate anion is not arranged in the core of the cavity.
:1 mole ratio yielded good quality single crystals of compound 2, which has the same space group as compound 1. Single-crystal X-ray diffraction analysis revealed that compound 2 is a coordination polymer constructed from closed molecular capsules. The asymmetric unit of compound 2 consists of two crystallographically distinct Sr2+ ions, a Q*[5] ligand, four nitrate anions, three coordinating water molecules and three uncoordinated water molecules. As shown in Fig. 3, Sr1 and Sr2 both have nine coordinate bonds. Each portal of the Q*[5] molecule is covered by one strontium cation, generating a completely closed molecular capsule {Sr2(H2O)3(NO3)[(NO3)⊂Q*[5]]}2+. The formation of the completely closed molecular capsule can ascribed to the size complementarity of the strontium cation and the Q*[5] portal. Interestingly, a nitrate anion is encapsulated in the cavity of the Q*[5] molecule. The distances between the nitrogen atom of the captured nitrate anion and the two strontium cations are 3.650 Å and 3.478 Å, respectively, indicating that the nitrate anion is not arranged in the core of the cavity.
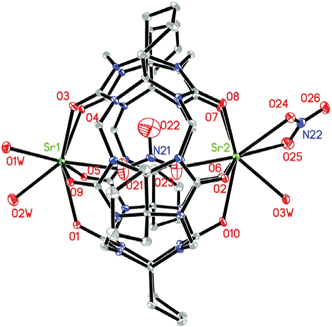 | ||
| Fig. 3 ORTEP diagram of compound 2 (asymmetric unit); displacement ellipsoids are drawn at the 30% probability level. Solvate water molecules and nitrate anions are omitted for clarity. | ||
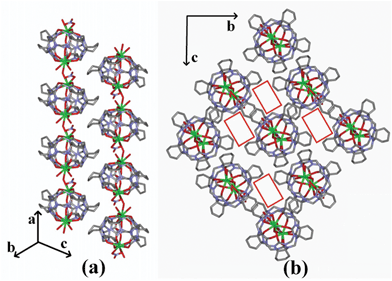
Owing to the bridging behavior of O3w, the adjacent {Sr2(H2O)3(NO3)[(NO3)⊂Q*[5]]}2+ molecule capsules are connected to each other, generating a linear polycationic chain {Sr2(H2O)3(NO3)[(NO3)⊂Q*[5]]}n2n+ along the a direction (Fig. 4a). It is worth mentioning that the adjacent chains are offset by half the repeat unit to maximize the interactions between the adjacent chains. In the crystal lattice of compound 2, three adjacent chains are connected through hydrogen-bonding interactions, producing a long channel along the a axis (Fig. 4b). The solvent accessible volume within the structure of compound 2 is 690.9 Å, which is 21.3% of the total unit cell volume.16
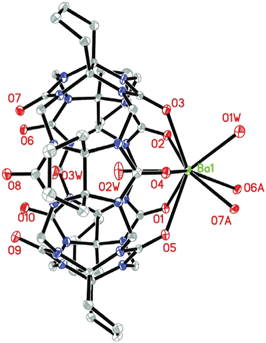 | ||
| Fig. 5 ORTEP diagram of compound 3 (asymmetric unit); displacement ellipsoids are drawn at the 30% probability level. Solvate water molecules and nitrate anions are omitted for clarity. | ||
The bridging behavior of the Ba2+ gives rise to the formation of a linear polycationic chain {Ba(H2O)[(H2O)2⊂Q*[5]]}n2n+ extending along the b axis (Fig. 6a). Measured along the equator of the Q*[5], the dihedral angle of the two neighboring molecular capsule is 37.92°. Similar to that in compound 2, the neighboring chains are offset by about one-half of the repeating unit along the opposite orientation. They form a two-dimensional close-packed layer on the ac plane (Fig. 6b). When viewing from the c axis, one can see many long wavelike gaps along the b axis (Fig. 6c), in which the nitrate anions and solvent waters were located.
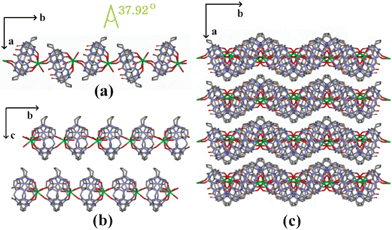 | ||
| Fig. 6 (a) 1D linear polycationic chain for compound 3 viewed down the c axis; (b) 2D close-packed layer of compound 3 viewed down the a axis; (c) packing diagram of compound 3 viewed down the c axis. O = red, C = grey, N = light blue and Ba = green. Water molecules and the nitrate anions are omitted for clarity. | ||
Comparison of the three Q*[5]-based molecular capsules
By reaction of the corresponding alkaline-earth metal salts with the Q*[5] ligand, three coordination architectures were prepared as expected, exhibiting similar coordination bonds of metal–OQ*[5] and similar molecule capsule structures. However, closer inspection reveals that significant differences exist among the three compounds. Firstly, compounds 1 and 2 have 1:2 ligand/metal compositions, while for 3, a 1:1 ligand/metal moiety was achieved although the three compounds were prepared using 1:3 ligand/metal mole ratios. Secondly, the calcium cations in compound 1 have eight coordinate bonds, while in compounds 2 and 3, the strontium and barium cations have nine. Thirdly, the calcium cations in compound 1 are almost coplanar with the five carbonyl oxygen atoms of the Q*[5] portal, whereas both the strontium and barium cations are out of the mean plane of the Q*[5] portal.
The most important difference among the three compounds is that they represent three different molecular capsule states. In compound 1, the Q*[5] portal is larger than the calcium cation. Thus, the Q*[5]-based molecular capsule is not closed completely even the Q*[5] molecule experiences some deformation. For compound 2, the radius of the strontium cation is slightly larger than the Q*[5] portal, and they form a completely closed molecular capsule. For compound 3, the barium ion can only cover one portal of the Q*[5] molecule and leans towards the portal of other Q*[5] molecule, generating an opened molecular capsule. Obviously, the radius (size) of metal ion is the underlying reason behind these differences.
Thermogravimetric analysis
Because compound 1 can be characterized as a dimer molecule, here we do not discuss its thermal stability. Since compounds 2 and 3 are structurally similar, compound 3 was selected for thermogravimetric analysis (TGA) to examine the thermal stability of these two compounds. The TGA study is performed under an air atmosphere. As can be seen in Fig. 7, compound 3 displays high thermal stability. The TGA curves clearly show that the uncoordinated water molecules are eliminated from the network (calcd 12.14%; found 12.10%) when the temperature is increased from room temperature to about 160 °C. No further weight loss was observed until the temperature reached 350 °C, which suggests that the polycationic chains of compound 3 are highly stable. On further heating, sharper weight loss is observed, suggesting that the polycationic chain has been broken down due to the decomposition of the Q*[5] ligands.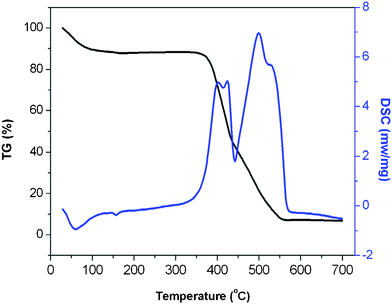 | ||
| Fig. 7 Thermogravimetric analysis diagram of compound 3. | ||
Conclusions
In summary, we have successfully prepared three structurally different compounds of Q*[5] ligand with alkaline-earth metal ions. Analysis of the crystallographic data of the three compounds clearly shows whether the molecular capsule is closed or opened and that the capsule is controlled by the radius (size) of the coordinated metal ions. In other words, the choice of metal ions is critical in controlling the molecular capsule states. Our present work not only provides an effective approach to manipulate the molecular capsule states, but also may be helpful for the design and construction of ionic radius-dependent coordination architectures.Experimental section
General
All reagents, such as Ca(NO3)2, Sr(NO3)2, Ba(NO3)2 and solvents employed were commercially available and used as received without further purification. The 1H NMR spectra were recorded at 20 °C on a Varian INOVA-400 spectrometer. The IR spectra were obtained on an Alpha Centaurt FT/IR spectrometer with KBr pellets in the 400–4000 cm−1 region. The C, H, and N microanalyses were carried out with a PE 240C elemental analyzer. The thermal gravimetric analyses (TGA) were carried out on a NETZSCH STA 449 C instrument under an air atmosphere with a heating rate of 10 °C min−1.Preparation of Q*5
Q*[5] was prepared according to the literature method.141H-NMR (D2O, 400 MHz): δ = 5.49 (d, 10H), 4.16 (d, 10H), 2.03 (s, 20H), 1.31 (s, 20H) ppm. Note that the 1H-NMR spectra of Q*[5] is slightly different from that in reported literature. IR (cm−1): 3540(s), 2954(m), 2060(m), 1743(s), 1650(m), 1467(s), 1411(s), 1373(s), 1355(s), 1335(s), 1314(s), 1270(s), 1213(s), 1173(s), 1032(w), 1014(m), 989(s), 906(m), 831(s), 764(s), 673(w), 593(w), 530(w), 462(w).Preparation of compounds
Crystal structure determination
Single-crystal X-ray data were collected on a Bruker Smart Apex CCD diffractometer. Standart graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) was employed. All data were collected in a 2θ range of 2.6° to 56° at 223 K. Absorption corrections were applied by using the multiscan program SADABS. All structures were solved by direct methods using SHELXS-97 and refine on F2 by full-matrix least-squares procedures with SHELXL-97.15 All non-hydrogen atoms were refined with anisotropic displacement parameters with the exception of disordered atoms. The carbon-bound hydrogen atoms were introduced at calculated positions. All hydrogen atoms were treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom. Analytical expressions of neutral-atom scattering factors were employed, and anomalous dispersion corrections were incorporated. For all compounds, no hydrogen atoms are given for all isolated and coordinated water molecules since it is difficult in cucurbituril compounds. Pertinent crystallographic data and refinement parameters for the three coordination compounds 1–3 are summarized in Table 1. Selected bond lengths (Å) for compounds 1–3 are shown in Table 2.| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C100H188N48O78Ca4 | C50H82N24O32Sr2 | C50H90N22O31Ba |
| M r | 3471.30 | 1706.64 | 1632.78 |
| Crystal system | Triclinic | Triclinic | Orthorhombic |
| Space group |
P |
P |
Pbca |
| a/Å | 12.7739(4) | 11.3443(7) | 23.864(9) |
| b/Å | 15.2460(5) | 15.8451(10) | 18.914(7) |
| c/Å | 19.3151(6) | 18.6881(12) | 29.501(11) |
| α/° | 82.9780(10) | 90.005(2) | 90 |
| β/° | 84.4020(10) | 97.736(2) | 90 |
| γ/° | 71.1360(10) | 103.096(2) | 90 |
| V/Å3 | 3526.14(19) | 3240.4(4) | 13316(8) |
| Z | 1 | 2 | 8 |
| Dc/g cm−3 | 1.635 | 1.749 | 1.629 |
| μ/mm−1 | 0.281 | 1.761 | 0.704 |
| Data/params | 13563/1060 |
12323/992 |
12765/938 |
| θ/° | 1.69–26.00 | 1.10–26.00 | 1.38–26.00 |
| GOF | 1.025 | 1.049 | 1.024 |
| Final R indices [I > 2σ (I)] | R 1 = 0.0571, wR2 = 0.1553 | R 1 = 0.0507, wR2 = 0.1355 | R 1 = 0.0561, wR2 = 0.1326 |
| R indices (all data) | R 1 = 0.0756, wR2 = 0.1724 | R 1 = 0.0749, wR2 = 0.1512 | R 1 = 0.0858, wR2 = 0.1489 |
| Symmetry transformations used to generate equivalent atoms: #1 = −x, −y, −z; #2 = x − 1, y, z; #3 = −x + 3/2, y + 1/2, z. | ||||
|---|---|---|---|---|
| 1 | Ca(1)–O(1) | 2.6319(14) | Ca(2)–O(6) | 2.6038(14) |
| Ca(1)–O(2) | 2.6069(15) | Ca(2)–O(7) | 2.4163(15) | |
| Ca(1)–O(3) | 2.4688(15) | Ca(2)–O(8) | 2.6764(16) | |
| Ca(1)–O(4) | 2.5164(14) | Ca(2)–O(9) | 2.4891(14) | |
| Ca(1)–O(5) | 2.4458(14) | Ca(2)–O(3w) | 2.3727(17) | |
| Ca(1)–O(1w) | 2.4103(17) | Ca(2)–O(4w) | 2.6023(16) | |
| Ca(1)–O(2w) | 2.3824(15) | Ca(2)–O(4w)#1 | 2.5590(15) | |
| 2 | Sr(1)–O(1) | 2.644(2) | Sr(2)–O(6) | 2.570(3) |
| Sr(1)–O(9) | 2.617(3) | Sr(2)–O(7) | 2.562(2) | |
| Sr(1)–O(3) | 2.660(2) | Sr(2)–O(8) | 2.663(2) | |
| Sr(1)–O(4) | 2.768(2) | Sr(2)–O(2) | 2.701(3) | |
| Sr(1)–O(5) | 2.570(3) | Sr(2)–O(10) | 2.710(2) | |
| Sr(1)–O(1w) | 2.534(3) | Sr(2)–O(3w) | 2.650(3) | |
| Sr(1)–O(2w) | 2.739(3) | Sr(1)–O(3w)#2 | 2.807(3) | |
| 3 | Ba(1)–O(1) | 2.836(3) | Ba(1)–O(6)#3 | 2.848(3) |
| Ba(1)–O(2) | 2.773(3) | Ba(1)–O(7)#3 | 2.684(3) | |
| Ba(1)–O(3) | 2.751(3) | Ba(1)–O(1w) | 2.772(4) | |
| Ba(1)–O(4) | 2.749(3) | Ba(1)–O(2w) | 2.835(4) | |
| Ba(1)–O(5) | 2.859(3) | |||
CCDC 856275–856277 contain the supplementary crystallographic data for compounds 1–3.†
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 20662003, 20961002 and 20971002), the Science and Technology Fund of Guizhou Province, China Postdoctoral Science Foundation (grant no. 20100481109), the International Collaborative Project of Guizhou Province (no. 2007400108) and the “Chun-Hui” Funds of Chinese Ministry of Education.References
- (a) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH: Weinheim, 1995 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley,Chichester, 2000 Search PubMed.
- (a) Reviews: L. M. Greig and D. Philp, Chem. Soc. Rev., 2001, 30, 287–302 RSC; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (c) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (d) J. P. Zhang and X. M. Chen, Chem. Commun., 2006, 1689–1699 RSC; (e) R. Robson, Dalton Trans., 2008, 5113 RSC; (f) T. Rehm and C. Schmuck, Chem. Commun., 2008, 801–813 RSC; (g) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (h) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC; (i) A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC; (j) M. Alajarin, R. A. Orenes, J. W. Steedc and A. Pastor, Chem. Commun., 2010, 46, 1394–1403 RSC.
- (a) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS; (b) D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS; (c) K. M. Fromm, Coord. Chem. Rev., 2008, 252, 856–885 CrossRef CAS; (d) K. S. Suslick, P. Bhyrappa, J.-H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283–291 CrossRef CAS; (e) B. Kesanli and W. Lin, Coord. Chem. Rev., 2003, 246, 305–326 CrossRef CAS; (f) H. L. Ngo and W. Lin, Top. Catal., 2005, 34, 85–92 CrossRef CAS; (g) H. Jiang and W. Lin, J. Am. Chem. Soc., 2006, 128, 11286–11297 CrossRef CAS; (h) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC; (i) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS.
- (a) Q. Ye, Y. M. Song, G. X. Wang, K. Chen, D. W. Fu, P. W. H. Chan, J. S. Zhu, S. D. Huang and R. G. Xiong, J. Am. Chem. Soc., 2006, 128, 6554–6555 CrossRef CAS; (b) M. Alvaro, E. Carbonell, B. Ferrer, F. Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS.
- (a) B. Li, R. J. Wei, J. Tao, R. B. Huang, L. S. Zheng and Z. P. Zheng, J. Am. Chem. Soc., 2010, 132, 1558–1559 CrossRef CAS; (b) D. W. Fu, W. Zhang and R. G. Xiong, Dalton Trans., 2008, 3946–3948 RSC; (c) M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K.-I. Sugiura, M. Yamashita, C. Coulon and R. Clerac, J. Am. Chem. Soc., 2005, 127, 3090–3099 CrossRef CAS.
- (a) C. D. Wu and W. B. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075–1078 CrossRef CAS; (b) D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916–920 CrossRef CAS; (c) C. D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS; (d) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS.
- (a) For example: B. Olenyuk, A. Fechtenkötter and P. J. Stang, J. Chem. Soc., Dalton Trans., 1998, 1707–1728 RSC; (b) B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS.
- (a) J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS; (b) A. I. Day, A. P. Arnold, R. J. Blanch and B. J. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS.
- (a) Reviews on cucurbit[n]uril: J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS; (b) J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS; (c) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (d) O. A. Gerasko, D. G. Samsonenko and V. P. Fedin, Russ. Chem. Rev., 2002, 71, 741–760 CrossRef CAS; (e) K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- (a) J. X. Liu, L. S. Long, R. B. Huang and L. S. Zheng, Cryst. Growth Des., 2006, 6, 2611–2614 CrossRef CAS; (b) J. X. Liu, L. S. Long, R. B. Huang and L. S. Zheng, Inorg. Chem., 2007, 46, 10168–10173 CrossRef CAS; (c) J. X. Liu, Y. F. Gu, R. L. Lin, W. R. Yao, X. H. Liu and J. Zhu, Supramol. Chem., 2010, 22, 130–134 CrossRef CAS; (d) J. X. Liu, Y. F. Hu, R. L. Lin, W. Q. Sun, X. H. Liu and W. R. Yao, J. Coord. Chem., 2010, 63, 1369–1378 CrossRef CAS; (e) X. L. Ni, J. X. Lin, Y. Y. Zheng, W. S. Wu, Y. Q. Zhang, S. F. Xue, Q. J. Zhu, Z. Tao and A. I. Day, Cryst. Growth Des., 2008, 8, 3446–3450 CrossRef CAS; (f) Y. Q. Zhang, J. P. Zeng, Q. J. Zhu, S. F. Xue and Z. Tao, J. Mol. Struct., 2009, 929, 167–173 CrossRef CAS.
- (a) P. Thuéry, Cryst. Growth Des., 2009, 9, 1208–1215 CrossRef; (b) P. Thuéry, Inorg. Chem., 2009, 48, 825–827 CrossRef; (c) P. Thuéry, Inorg. Chem., 2009, 48, 4497–4513 CrossRef; (d) P. Thuéry, Inorg. Chem., 2010, 49, 9078–9085 CrossRef.
- (a) O. A. Geras'ko, E. A. Mainicheva, M. I. Naumova, M. Neumaier, M. M. Kappes, S. Lebedkin, D. Fenske and V. P. Fedin, Inorg. Chem., 2008, 47, 8869–8880 CrossRef CAS; (b) O. A.Geras'ko, E. A. Mainicheva, M. I. Naumova, O. P. Yurjeva, A. Alberola, C. Vicent, R. Llusar and V. P. Fedin, Eur. J. Inorg. Chem., 2008, 416–424 Search PubMed; (c) D. G. Samsonenko, J. Lipkowski, O. A. Gerasko, A. V. Virovets, M. N. Sokolov, V. P. Fedin, J. G. Platas, R. Hernandez-Molina and A. Mederos, Eur. J. Inorg. Chem., 2002, 2380–2388 CrossRef CAS.
- (a) W. A. Freeman, W. L. Mock and N.-Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367–7368 CrossRef CAS; (b) W. A. Freeman, Acta Crystallogr., Sect. B, 1984, 40, 382–387 CrossRef; (c) Y. M. Jeon, J. Kim, D. Whang and K. Kim, J. Am. Chem. Soc., 1996, 118, 9790–9791 CrossRef CAS; (d) J. Heo, J. Kim, D. Whang and K. Kim, Inorg. Chim. Acta, 2000, 297, 307–312 CrossRef CAS; (e) J. Heo, S. Y. Kim, D. Whang and K. Kim, Angew. Chem., Int. Ed., 1999, 38, 641–643 CrossRef CAS; (f) D. Whang, J. Heo, J. H. Park and K. Kim, Angew. Chem., Int. Ed., 1998, 37, 78–80 CrossRef CAS.
- J. Z. Zhao, H.-J. Kim, J. Oh, S. Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233–4235 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef; (b) G. M. Sheldrick, SHELXL-97 Program for the Solution and Refinement of Crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1998 Search PubMed.
Footnote |
| † CCDC 856275–856277 for the compounds 1–3. For crystallographic data in CIF and other electronic format See DOI: 10.1039/c2ra20569f/. |
| This journal is © The Royal Society of Chemistry 2012 |