Hierarchical magnetic iron (III) oxides prepared by solid-state thermal decomposition of coordination polymers†
Ming
Hu
a,
Ji-Sen
Jiang
*a,
Fan-Xing
Bu
a,
Xun-Liang
Cheng
a,
Chu-Cheng
Lin
b and
Yi
Zeng
b
aDepartment of Physics, Center of Functional Nanomaterials and Devices, East China Normal University, 200241, Shanghai, China. E-mail: jsjiang@phy.ecnu.edu.cn
bShanghai Institute of Ceramics, Chinese Academy of Science, Shanghai, China
First published on 16th April 2012
Abstract
By calcining Prussian Blue (PB) microcrystals with well-defined morphologies at different temperatures (300 °C, 450 °C and 600 °C, respectively) in air, β-Fe2O3, γ-Fe2O3 and α-Fe2O3 with different shapes were successfully prepared. It was found that the heating rate played a critical role in the shape and hierarchical structure of the prepared iron(III) oxides. When the heating rate was as high as 10 °C min−1, hierarchical iron(III) oxides could be obtained, while when the heating rate was as low as 1 °C min−1, iron(III) oxides with smooth surfaces could be obtained. The hierarchical iron(III) oxides showed an interesting magnetic response at room temperature. The differences of magnetic properties for the hierarchical iron(III) oxides prepared at different temperatures should be caused by the composition of different phases.
1. Introduction
Iron(III) oxides are very important classes of metal oxides, which have been applied in many fields, such as magnetic records, catalysis, and batteries etc.1 In general, there are four kinds of phase of iron(III) oxides including α-Fe2O3, β-Fe2O3, γ-Fe2O3, and ε-Fe2O3. Among them, α-Fe2O3 and γ-Fe2O3 are the most common phases, while pure β-Fe2O3 and ε-Fe2O3 are still rare.2 As the unique properties of iron(III) oxides are determined by their crystal structures, it is of particular interest to get different phases of iron(III) oxides. To date, many methods have been developed to fabricate iron(III) oxides,3–5 among which the most effective way to get rare polymorphs of iron(III) oxides is solid-state decomposition of iron-containing salts.1e,5b–d Because of the different stabilization of four polymorphs of iron(III) oxides, different polymorphs could be easily obtained at different calcination temperatures, by changing other thermal decomposition parameters or tuning the particle sizes.5b–d For example, the phase transformation of γ → ε→ β → α was observed by tuning the particles sizes.5cBenefiting from the development of nanotechnology, shape-related properties of materials have been proved in many fields.6 Inspired by that, research on shape-controlled synthesis of iron(III) oxides has also become very hot in the past a few years. Hierarchical iron oxides, which are composed of small components (for example, nanoparticles, nanorods etc.), offer enhanced performances in many fields, such as lithium ion batteries, solar cells, water treatment catalysis and sensing etc.1f,g,7 Therefore, to fabricate hierarchical iron(III) oxides is of both fundamental and practical importance. To date, great effort has been made, however, most of the methods were based on a wet-chemical reaction.4a,7a,b,d,f,g,I,j In general, wet-chemical methods are not effective in getting multiple phases of iron(III) oxides. Thus, developing a solid-state method to fabricate hierarchical iron(III) oxides with different phases will be very interesting and offer the opportunity to get multi-functional materials. Fortunately, there are already a few reports about the solid-state shape-controlled synthesis of iron(III) oxides,1e,2a,5b–d such as nanoparticles with highly catalytic activity. However, there is still lack of fabrication of hierarchical iron(III) oxides via solid-state decomposition. 8
Coordination polymers, which are composed of metal ions and organic groups, are very attractive materials in separation, gas adsorption and catalysis etc.9 When calcined at high temperature in air, the organic part of the coordination polymers will be easily removed, which makes coordination polymers promising precursor to fabricate metal oxides. In recent years, coordination polymers with well-defined shapes were utilized as precursor to prepare porous or non-porous metal oxides by thermal decomposition.5b,10,11 Among these coordination polymers, Prussian Blue (PB, Fe4[Fe(CN)6·14H2O) is very suitable for fabricating iron(III) oxides. Since the pioneering work of Zbořil et al. on the thermal decomposition of PB,5b iron(III) oxides with different phases have been fabricated.11 For example, by using well-defined PB microcrystals or nanofilms, mesoporous iron(III) oxides were fabricated.11 In this paper, we selected PB microcrystals as precursors, and successfully transformed them into hierarchical iron(III) oxides microcrystals. Such a route opens a facile route to fabricate iron(III) oxides with exquisite structure, and enriches the solid-state decomposition of PB family.
2. Experimental
Synthesis of elongated PB microcrystals11a
In a typical synthesis, 168.9 mg K4[Fe(CN)6] and 291.6 mg cetyltrimethylammonium bromide (CTAB) were mixed into 13 mL distilled water under stirring, then 20 mL isopropanol and 7 mL 36% hydrochloric acid were added into the mixture in sequence, finally the mixture became transparent. The transparent solution were transferred into a 50 mL Teflon-lined stainless-steel autoclave, sealed and maintained at 120 °C for 24 h. After the solution was cooled to room temperature, the obtained deep blue solid was collected by centrifugation, washed several times with water and ethanol, and then dried in a vacuum oven at 60 °C for 10 h.Synthesis of truncated PB mesocrystals12
In a typical synthesis, 236.6 mg K4[Fe(CN)6] and 500 mg polyethylene glycol 4000 (PEG 4000) were mixed into 29 mL distilled water under stirring, then 6 mL 36% hydrochloric acid was added into the solution under stirring. 10 min later, the mixture was transferred into a 45 mL Teflon-lined stainless-steel autoclave, sealed and maintained at 120 °C for 20 h. After the solution was cooled to room temperature, the obtained deep blue solid was collected by centrifugation, washed several times with water and acetone, and then dried in a vacuum oven at 60 °C for 10 h.Preparation of hierarchical iron oxides
Thermal treatment of PB was conducted using an electric muffle furnace in air. In a typical process, 40 mg PB powder prepared as above was put into a melting pot. Then, the melting pot was put inside a muffle furnace and heated to a set temperature under constant heating rate. After heated at the set temperature for 30 min, the sample was kept inside the furnace, and cooled naturally. The resulted product was collected for subsequent characterization.Characterization
The phase composition of samples was monitored by X-ray diffraction (XRD) using an X'Pert-Pro MPD diffractometer with Cu-Kα radiation and conventional θ–2θ geometry. Scanning electron microscopy (SEM) measurements were made on a JSM 6700F microscope. The room-temperature magnetic hysteresis loop of the iron(III) oxides was measured by a vibrating sample magnetometer (HH-15, China).3. Results and discussion
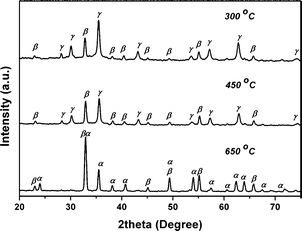
For solid-state transformations, calcination temperature is a critical parameter.5b–d To investigate the impact of temperature on the thermal decomposition of PB microcrystals, we selected elongated PB microcrystals as precursor (See Fig. S1 in Supplementary Information for SEM image†). The elongated PB microcrystals were calcined at different temperatures with a heating rate of 10 °C min−1. Fig. 1 shows the XRD patterns of products obtained at various temperatures. When calcined at 300 °C, the products were mixed of β-Fe2O3 (JCPDS card 39-0238) and γ-Fe2O3 (JCPDS card 39-1346). When the temperature was elevated to 450 °C, the products were still composed of β-Fe2O3 and γ-Fe2O3. When the temperature was as high as 650 °C, most of the β-Fe2O3 and γ-Fe2O3 were converted into α-Fe2O3 (JCPDS card 79-1741). However, very weak peaks of β-Fe2O3 and γ-Fe2O3 could still be observed, which suggested the existence of a trace amount of impurities. On the basis of the intensity of (104) and (110), the phase ratio of each sample was calculated and listed in Table 1. Clearly, we could see a temperature-dependent phase change. For the polymorphs of iron(III) oxides, the stabilities of different phases are different, which offers the possibility for such phase variations.5b–c As PB was calcined in air, the cyanide group was decomposed and oxidized, and released from the PB crystals. Subsequently, the remaining iron elements were oxidized into iron(III) oxides by oxygen in air. Because β-Fe2O3 and γ-Fe2O3 were easily generated at a low temperature, the thermal-decomposition of PB at 300 °C and 450 °C could result in β-Fe2O3 and γ-Fe2O3.5b,11a However, as β-Fe2O3 and γ-Fe2O3 were less stable than α-Fe2O3, they were easily transformed into α-Fe2O3 at higher temperature.2a,5b Therefore, we got β-Fe2O3 and γ-Fe2O3 at a lower temperature (300 and 450 °C), and got α-Fe2O3 at a higher temperature (650 °C). Such temperature-dependent phase changes indicate that it is easy to get different phase of iron(III) oxides by tuning the calcining temperature. | ||
| Fig. 1 XRD patterns of products obtained by calcining elongated PB microcrystals at various temperatures under 10 °C min−1. | ||
| Heating Rate (°C min−1) | Calcination T/°C | α phase | β phase | γ phase |
|---|---|---|---|---|
| 10 | 300 | None | 17 wt% 60 nm | 83 wt% 43 nm |
| 10 | 450 | None | 21 wt% 80 nm | 79 wt% 62 nm |
| 10 | 650 | 93 wt% 120 nm | 5 wt% 110 nm | 2 wt% 115 nm |
| 1 | 300 | None | 31 wt% | 69 wt% |
| 1 | 450 | None | 51wt% | 49 wt% |
| 1 | 650 | 60 wt% | 40 wt% | None |
To evaluate the morphology of these obtained iron(III) oxides, SEM images are shown in Fig. 2. The initial morphology of the PB precursor was roughly retained. However, all the samples exhibited rough surfaces. Enlarged images taken from the marked areas indicate the larger crystals are indeed constituted of nanocrystals, which suggests these iron(III) oxides are all hierarchical structures. The sizes of the nanocrystals varies from several tens of nanometers to more than one hundred nanometers, which depends on the calcination temperature. Such a result is also consistent with the crystallite sizes calculated by Scherrer formula based on the XRD patterns in Fig. 1, which were listed in Table 1. Such variation of sizes indicates a coarsening of grains which may be caused by fusing of nanoparticles at high temperature.
 | ||
| Fig. 2 SEM images of products obtained by calcining elongated PB microcrystals at various temperatures under 10 °C min−1. (a) and (b) 300 °C. (c) and (d) 450 °C. (e) and (f) 650 °C. | ||
To understand whether heating rate can influence the phase composition of iron(III) oxides, and understand the mechanism for the formation of hierarchical structures, we have calcined PB microcrystals under a lower heating rate of 1 °C min−1 as a contrast.
Compared to the samples obtained at higher heating rate of 10 °C min−1 (Fig. 2), the samples obtained under a lower heating rate present different structures and phases, as shown in Fig. 3, Fig. 4 and Table 1. The low heating rate obviously favored the generation of β-Fe2O3. Both the amount of γ-Fe2O3 and α-Fe2O3 were reduced. While for the microstructure, smooth surfaces were observed after calcining at all temperatures. Although hierarchical structure could also be obtained at 650 °C, the worm-like nanostructures are larger than the particle nanostructures, which indicates nanoparticle fusion happened under slow heating.8a Obviously, the heating rate plays an important role in the shape variation of iron(III) oxides obtained by calcining. The higher heating rate could promote the formation of hierarchical structure, while the lower heating rate could prevent the formation of hierarchical structure.
 | ||
| Fig. 3 SEM images of products obtained by calcining elongated PB microcrystals at various temperatures under 1 °C min−1. (a) and (b) 300 °C. (c) and (d) 450 °C. (e) and (f) 650 °C | ||
 | ||
| Fig. 4 XRD patterns of products obtained by calcining elongated PB microcrystals at various temperatures under 1 °C min−1. | ||
When crystallization happens in solution, the large excess of supersaturated species usually lead to explosive formation of a large amount of nuclei, and of course leads to nanocrystals with small sizes due to the lack of species for consumption by the growth of so many nuclei.13 In contrast, when the supersaturation degree is low, fewer nuclei can be formed. As a result, there are enough species to attach to the nuclei to make them grow into large crystals. In our case, although it is a solid-state reaction, the formation of hierarchical structure is still somehow like crystallization in solution. During the conversion process of PB, the ferrous iron(II) is gradually oxidized into iron(III) oxide due to the time-consuming thermal-decomposition process of PB. A higher heating rate could accelerate the process, thus leading to drastic formation of iron oxides nuclei. As there are not enough species to make the nuclei grow large, only nanoparticles could be obtained. When the heating rate was low, fewer nuclei formed at first, thus the subsequently formed iron oxides could allow the growth of preformed nuclei and finally resulted in crystals with smooth surfaces.
As a higher heating rate favors the generation of hierarchical iron oxides, it is very interesting to know whether this phenomenon will happen to other PB crystals. Due to the symmetric shape and smooth surfaces of elongated PB microcrystals, it is more like a single crystal. In recent years, another kind of crystals, mesocrystals, have drawn much attention. Mesocrystals represent a group of crystals which formed by self-assembly or self-aggregation of small particles (e.g. nanocrystals) in an oriented way.14 Owing to the specific structure, mesocrystals show interesting properties different from single crystals.15 Therefore, PB mesocrystals may be an ideal choice to investigate whether rapid heating could be a general route for fabricating hierarchical structure. Here we chose truncated PB mesocrystals as a precursor for thermal decomposition.12 Such mesocrystals show a very close shape compared to elongated PB microcrystals, while the surfaces are curved and rough, which comes from the mesocrystalline structure (Fig. S2†). Fig. 5 and 6 illustrate the products calcined in air under heating rates of 10 °C min−1 and 1 °C min−1, respectively. Similar to PB single crystals, hierarchical structures were successfully obtained under a higher heating rate while lower heating rate prevented the generation of hierarchical structures. Such a phenomenon strongly suggests that heating rate plays a critical role in the fabrication of hierarchical iron oxides, and the thermal decomposition of PB microcrystals could be a general route. In addition, it is worth noting that the nanoparticles which formed the hierarchical structures shown in Fig. 5 are much smaller than the nanoparticles shown in Fig. 2. Considering that PB mesocrystals are composed of nanoparticles, large number of crystal boundaries surely exist in a mesocrystal. When the nucleation of iron oxides started, the growth of iron oxides was confined by the pre-existing crystal boundaries of mesocrystals, which finally resulted in hierarchical structures composed of smaller particles.
 | ||
| Fig. 5 SEM images of products obtained by calcining truncated PB mesocrystals at various temperatures under 10 °C min−1. (a) and (b) 300 °C. (c) and (d) 450 °C. (e) and (f) 650 °C. | ||
 | ||
| Fig. 6 SEM images of products obtained by calcining truncated PB mesocrystals at various temperatures under 1 °C min−1. (a) and (b) 300 °C. (c) and (d) 450 °C. (e) and (f) 650 °C. | ||
As iron(III) oxides are important magnetic materials, the magnetic properties of the hierarchical iron(III) oxides were evaluated. The room-temperature magnetization hysteresis curves shown in Fig. 7 indicate the magnetic response decreased with the increase of calcining temperature. The products shown in Fig. 2a and 2c present significant ferromagnetic behaviors, while the samples shown in Fig. 2e present weak ferromagnetic behavior. Such differences are consistent with the composition of these samples. The samples calcined at 300 °C and 450 °C are mixed β-Fe2O3 and γ-Fe2O3 while the sample calcined at 650 °C is almost pure α-Fe2O3. Among the three phases of iron(III) oxides, β-Fe2O3 is paramagnetic, α-Fe2O3 shows very weak ferromagnetic behavior at room-temperature, only γ-Fe2O3 shows a strong magnetic response because of its ferromagnetic properties.2a Therefore, the main contributor of the strong magnetic response of the samples calcined at 300 °C and 450 °C should be γ-Fe2O3. The weak ferromagnetic response should come from α-Fe2O3 itself and trace amounts of γ-Fe2O3. The differences of the values of saturated magnetization between the samples calcined at 300 °C and 450 °C indicate the amount of γ-Fe2O3 in both samples is different.
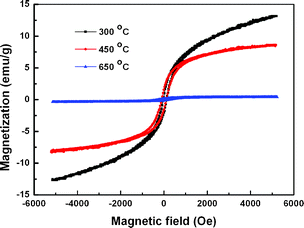 | ||
| Fig. 7 Magnetic hysteresis curves of products obtained by calcining elongated PB microcrystals at various temperatures under 10 °C min−1. | ||
4. Conclusions
In conclusion, we successfully prepared hierarchical iron(III) oxides by calcination of PB microcrystals. By tuning the calcination temperature from 300 °C to 650 °C, the composition of iron(III) oxides could be changed from β-Fe2O3 and γ-Fe2O3 to α-Fe2O3. In addition, it was found that when the heating rate was as high as 10 °C min−1, the products were hierarchical structures. These iron(III) oxides with hierarchical structures showed magnetic response at room temperature. Considering the hierarchical structure and magnetic properties, these iron(III) oxides were promising materials in catalysis and lithium ion batteries etc. Moreover, such a solid-state process would be a general route to fabricate many other hierarchical metal oxides by choosing appropriate coordination polymers containing different metal atoms.Acknowledgements
This research project was supported by the National Natural Science Foundation of China (Grant No. 21173084), the PhD Program Scholarship Fund of ECNU 2008 (Grant 20080045) and Equipment Sharing Platform of ECNU.References
- (a) R. Dronskowski, Adv. Funct. Mater., 2001, 11, 27–29 CrossRef CAS; (b) N. Zhao, W. Ma, Z. Cui, W. Song, C. Xu and M. Gao, ACS Nano, 2009, 3, 1775–1780 CrossRef CAS; (c) F. Wang, Y. Xu, D. I. C. Wang and Z. Li, J. Am. Chem. Soc., 2009, 131, 12892–12893 CrossRef; (d) F. Shi, M. K. Tse, M. M. Pohl, A. Brückner, S. Zhang and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 8866–8868 CrossRef CAS; (e) M. Hermanek, R. Zboril, I. Medrik, J. Pechousek and C. Gregor, J. Am. Chem. Soc., 2007, 129, 10929–10936 CrossRef CAS; (f) B. Sun, J. Horvat, H. S. Kim, W.–S. Kim, J. Ahn and G. Wang, J. Phys. Chem. C, 2010, 114, 18753–18761 CrossRef CAS; (g) K. Brezesinski, J. Haetge, J. Wang, S. Mascotto, C. Reitz, A. Rein, S. H. Tolbert, J. Perlich, B. Dunn and T. Brezesinski, Small, 2011, 7, 407–414 CrossRef CAS; (h) E. Thimsen, F. L. Formal, M. Grätzel and S. C. Warren, Nano Lett., 2011, 11, 35–43 CrossRef CAS.
- (a) R. Zboril, M. Mashlan and D. Petridis, Chem. Mater., 2002, 14, 969–982 CrossRef CAS; (b) J. Tuček, R. Zbořil, A. Namai and S. -I. Ohkoshi, Chem. Mater., 2010, 22, 6483–6505 CrossRef.
- Y. Kusano, T. Fujii, J. Takada, M. Fukuhara, A. Doi, Y. Ikeda and M. Takano, Chem. Mater., 2008, 20, 151–156 CrossRef CAS.
- (a) M. Cao, T. Liu, S. Gao, G. Sun, X. Wu, C. Hu and Z. L. Wang, Angew. Chem., Int. Ed., 2005, 44, 4197–4201 CrossRef CAS; (b) X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS; (c) C. -J. Jia, L. -D. Sun, Z. -G. Yan, L. -P. You, F. Luo, X. -D. Han, Y. -C. Pang, Z. Zhang and C. -H. Yan, Angew. Chem., Int. Ed., 2005, 44, 4328–4333 CrossRef CAS; (d) Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329–6332 CrossRef CAS.
- (a) E. Taboada, M. Gich and A. Roig, ACS Nano, 2009, 3, 3377–3382 CrossRef CAS; (b) R. Zboril, L. Machala, M. Mashlan and V. Sharma, Cryst. Growth Des., 2004, 4, 1317–1325 CrossRef CAS; (c) S. Sakurai, A. Namai, K. Hashimoto and S. -I. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 18299–18303 CrossRef CAS; (d) M. Hermanek and R. Zboril, Chem. Mater., 2008, 20, 5284–5295 CrossRef CAS.
- (a) D. Xu, Z. Liu, H. Yang, Q. Liu, J. Zhang, J. Fang, S. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217–4221 CrossRef CAS; (b) Q. Song and Z. J. Zhang, J. Am. Chem. Soc., 2004, 126, 6164–6168 CrossRef CAS; (c) Y. X. Chen, S. P. Chen, Z. Y. Zhou, N. Tian, Y. X. Jiang and S. G. Sun, J. Am. Chem. Soc., 2009, 131, 10860–10862 CrossRef CAS; (d) X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS; (e) K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS; (f) H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–642 CrossRef CAS.
- (a) Z. An, J. Zhang, S. Pan and F. Yu, J. Phys. Chem. C, 2009, 113, 8092–8096 CrossRef CAS; (b) S. Zeng, K. Tang, T. Li, Z. Liang, D. Wang, Y. Wang, Y. Qi and W. Zhou, J. Phys. Chem. C, 2008, 112, 4836–4843 CrossRef CAS; (c) A. Kay, I. Cesar and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 15714–15721 CrossRef CAS; (d) S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048–3055 CrossRef CAS; (e) S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS; (f) R. L. Spray and K. S. Choi, Chem. Mater., 2009, 21, 3701–3709 CrossRef CAS; (g) S. W. Cao and Y. J. Zhu, J. Phys. Chem. C, 2008, 112, 6253–6257 CrossRef CAS; (h) X. Gou, G. Wang, X. Kong, D. Wexler, J. Horvat, J. Yang and J. Park, Chem.–Eur. J., 2008, 14, 5996–6002 CrossRef CAS; (i) D. Mao, J. Yao, X. Lai, M. Yang, J. Du and D. Wang, Small, 2011, 7, 578–582 CrossRef CAS; (j) X. L. Fang, C. Chen, M. S. Jin, Q. Kuang, Z. X. Xie, S. Y. Xie, R. B. Huang and L. S. Zheng, J. Mater. Chem., 2009, 19, 6154–6160 RSC.
- (a) R. Amutha, M. Muruganandham, M. Sathish, S. Akilandeswari, R. P. S. Suri, E. Repo and M. Sillanpää, J. Phys. Chem. C, 2011, 115, 6367–6374 CrossRef CAS; (b) M. Muruganandham, R. Amutha, B. Ahmmad, E. Repo and M. Sillanpää, J. Phys. Chem. C, 2010, 114, 22493–22501 CrossRef CAS.
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (c) X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS; (d) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS; (e) O. K. Farha, A. Ö. Yazaydin, I. Eryazici1, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS.
- (a) S. Jung, W. Cho, H. J. Lee and M. Oh, Angew. Chem., Int. Ed., 2009, 48, 1459–1462 CrossRef CAS; (b) W. Cho, S. Park and M. Oh, Chem. Commun., 2011, 47, 4138–4140 RSC; (c) W. Cho, Y. H. Lee, H. J. Lee and M. Oh, Chem. Commun., 2009, 4756–4758 RSC; (d) C. M. Burgess, N. Yaob and A. B. Bocarsly, J. Mater. Chem., 2009, 19, 8846–8855 RSC; (e) W. Cho, Y. H. Lee, H. J. Lee and M. Oh, Adv. Mater., 2011, 23, 1720–1723 CrossRef CAS; (f) L. L. Li, J. Xu, Q. Yuan, Z. X. Li, W. G. Song and C. H. Yan, Small, 2009, 5, 2730–2737 CrossRef CAS.
- (a) M. Hu, J. S. Jiang and Y. Zeng, Chem. Commun., 2010, 46, 1133–1135 RSC; (b) X. Roy, J. K. H. Hui, M. Rabnawaz, G. Liu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2011, 50, 1597–1602 CrossRef CAS.
- M. Hu, J. S. Jiang, C. C. Lin and Y. Zeng, CrystEngComm, 2010, 12, 2679–2683 RSC.
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- (a) H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef; (b) H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350–2365 CrossRef.
- X. L. Wu, S. J. Xiong, Z. Liu, J. Chen, J. C. Shen, T. H. Li, P. H. Wu and P. K. Chu, Nat. Nanotechnol., 2011, 6, 103–106 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: SEM images of PB microcrystals. See DOI: 10.1039/c2ra01190e/ |
| This journal is © The Royal Society of Chemistry 2012 |