Controlled hydrogenation of aromatic compounds by platinum nanowire catalysts†
Zhiqiang
Guo
a,
Lei
Hu
a,
Hsiao-hua
Yu
*b,
Xueqin
Cao
*ac and
Hongwei
Gu
*a
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, P. R. China. E-mail: hongwei@suda.edu.cn; xqcao@suda.edu.cn
bInitiative Research Unit, RIKEN Advanced Science Institute, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: bruceyu@riken.jp
cNational Engineering Laboratory for Modern Silk, Soochow University, Suzhou, Jiangsu Province 215123, P. R. China
First published on 29th February 2012
Abstract
Aromatic hydrogenation to form cyclohexane derivatives is one of the most important steps in both the petrochemical industry, for clean diesel fuel generation, and the pharmaceutical industry, for safe drug synthesis. We report herein, pure ultra-thin Pt nanowire catalysts with no supporting matrix and their high activities towards the hydrogenation of aromatic compounds (>99% conversion, 1 MPa initial hydrogen pressure, 70 °C). The catalysts can also be recycled up to 40 times with no loss in activity.
Introduction
The efficient synthesis of cyclohexane derivatives via the catalytic hydrogenation of aromatic compounds is of both scientific and industrial importance. However, the complete hydrogenation of aromatics still requires highly energetic (>100 °C) and dangerous reaction conditions (50 atm H2) due to the lack of efficient catalysts. Recent publications suggest that palladium,1 ruthenium,2 rhodium,3 iridium4 and nickel5 may be used to achieve the hydrogenation of aromatic compounds under mild conditions. The groups of Jiang and Han6 recently reported a selective phenol hydrogenation over a supported Pd-Lewis acid catalyst at 30 °C and 1 MPa H2. Saim Özkar7 and coworkers reported the complete hydrogenation of neat benzene using ruthenium(0) nanoclusters as the catalyst under mild reaction conditions. The Anderson group8 reported a highly active and selective hydrogenation of phenyl compounds bearing polar functional groups, such as acids and amides, over a palladium catalyst. However, the utilization of platinum-based catalysts has been less investigated, even though platinum (Pt) is commonly regarded as one of the most active catalysts for the hydrogenation of unsaturated bonds. Somorjai and Yang9 reported the catalytic hydrogenation of pyrrole using Pt nanocrystals and found that the activity and selectivity depended heavily on the shape and size of nanocrystals. A similar phenomenon was also observed by the Han group10 using palladium nanocrystals.Recently, one-dimensional Pt nanowires (Pt NWs) have been successfully synthesized using a de-alloy process. These synthesized Pt NWs lead to high conversions in nitro group reductions under 1 atm initial hydrogen pressure.11 Herein, we report the more challenging and versatile hydrogenation of aromatic compounds using ultra-thin Pt NWs. Outstanding catalytic activities and selectivities have been demonstrated at low temperatures (60–70 °C) and low initial hydrogen pressures (1 MPa) in the presence of a Lewis acid. Moreover, the Pt NWs can be recycled via centrifugation and reused for 40 cycles with no loss in activity.
Results and discussion
Initially, alkenes and alkynes were examined as substrates in the catalytic hydrogenation. We observed the formation of fully saturated alkanes from the corresponding unsaturated compounds within 1–2 h (ESI, Table S1†). Styrene was fully converted to ethylbenzene in 90 min and 10 min, at initial hydrogen pressures of 1 atm and 4 atm at 40 °C. The TOF values were calculated as 1333 h−1 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, which are better than any previously reported data to our knowledge (ESI, Table S2 and Table S3†).12 The initial tests demonstrated high catalytic activities for the Pt NWs in the hydrogenation of unsaturated carbon bonds. Higher initial hydrogen pressures increased the catalytic activity, resulting in faster reactions.
000 h−1, which are better than any previously reported data to our knowledge (ESI, Table S2 and Table S3†).12 The initial tests demonstrated high catalytic activities for the Pt NWs in the hydrogenation of unsaturated carbon bonds. Higher initial hydrogen pressures increased the catalytic activity, resulting in faster reactions.
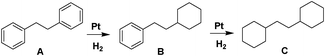
Encouraged by the initial results, we selected 1,2-diphenylethane (A) as our model compound to further explore the Pt NW catalyzed aromatic hydrogenation reaction. Isopropanol (IPA) was first selected as the solvent with a reaction temperature of 60 °C. However, the conversion of A was only 11.8% (Table 1, entry 1) and partially hydrogenated product, 1-(2-cyclohexylethyl)benzene (B), was the major product. We then switched the solvent to acetic acid (HOAc) and the conversion of A was dramatically increased to 58.5%. The levels of fully hydrogenated product (C) were also increased from 93.4:6.6 (B:C) to 75.7:24.3 (Table 1, entry 2). To increase the hydrogenation efficiency, we assessed the addition of Lewis acids, which proved effective activators of the aromatic compounds.5,13 As shown in Table 1, entry 3, 95.9% of A was converted and the fully hydrogenated C was the major product when AlCl3 (0.25 mmol) was employed. We found that utilization of other Lewis acids (FeCl3, ZnCl2) did not enhance the activity of the Pt NWs (Table 1, entries 4 and 5) and the conversions were only 62% and 42.3%, respectively, with B as the major product. Compared to other solvents (IPA, ethanol and methanol), reactions carried out in HOAc resulted in the highest yields for the fully hydrogenated product (ESI, Table S4†). Increasing or reducing the molar ratio of AlCl3 had little effect on the catalytic hydrogenation (Table 1, entries 7 and 8). On the other hand, higher reaction temperatures accelerated the reaction. At 70 °C, >99% conversion was achieved with the selectivity for C exceeding 99% within 25 h at 1 MPa initial hydrogen pressure (Table 1, entry 6). The initial hydrogen pressure also influences the reaction rate. At 0.5 MPa initial hydrogen pressure, the conversion of A after 25 h was only 11.7% (Table 1, entry 9). Conversely, at 2 MPa initial hydrogen pressure, A was fully converted to C within 22 h. (Table 1, entry 10)
| Entry | T (°C) | t (h) | Solvent | Cocat. | Conv. (%) | Select. (%) | |
|---|---|---|---|---|---|---|---|
| B | C | ||||||
| a Reaction conditions: Pt nanowires (0.005 mmol), 1,2-diphenylethane (1.0 mmol), solvent (2 mL), Lewis acid (0.25 mmol) and hydrogen pressure (1 MPa). b 0.5 mmol AlCl3. c 0.125 mmol AlCl3. d 0.5 MPa initial hydrogen pressure. e 2 MPa initial hydrogen pressure. The conversion and selectivity were determined by gas chromatography (GC). IPA = Isopropanol, HOAc = Acetic Acid. | |||||||
| 1 | 60 | 25 | IPA | — | 11.8 | 93.4 | 6.6 |
| 2 | 60 | 25 | HOAc | — | 58.5 | 75.7 | 24.3 |
| 3 | 60 | 25 | HOAc | AlCl3 | 95.5 | 33.0 | 67.0 |
| 4 | 60 | 25 | HOAc | FeCl3 | 62.0 | 54.6 | 45.4 |
| 5 | 60 | 25 | HOAc | ZnCl2 | 42.3 | 69.1 | 30.9 |
| 6 | 70 | 25 | HOAc | AlCl3 | 100 | 0.6 | 99.4 |
| 7b | 70 | 25 | HOAc | AlCl3 | 97.0 | 31.1 | 68.9 |
| 8c | 70 | 25 | HOAc | AlCl3 | 91.2 | 44.0 | 56.0 |
| 9d | 70 | 25 | HOAc | AlCl3 | 11.7 | 70.7 | 29.3 |
| 10e | 70 | 22 | HOAc | AlCl3 | 100 | 0.3 | 99.7 |
| 11 | 70 | 5 | HOAc | AlCl3 | 46.8 | 73.9 | 26.1 |
| 12 | 70 | 10 | HOAc | AlCl3 | 53.5 | 74.4 | 25.6 |
| 13 | 70 | 15 | HOAc | AlCl3 | 75.4 | 57.5 | 42.5 |
| 14 | 70 | 22 | HOAc | AlCl3 | 87.9 | 45.8 | 54.2 |
The catalytic hydrogenation process was also followed by gas chromatography (GC), as shown in Fig. 1A for the conversion of A to B and B to C. These two hydrogenation processes were executed in parallel. The selectivity for C increases with increasing conversion of A (Table 1, entries 6 and 11–14). This observation is also supported by time-dependent proton NMR analysis (ESI, Fig. S1†). The signal for the –CH2– adjacent to the phenyl group decreases, while that for the –CH2– adjacent to the cyclohexyl increases. The partial reduction of the benzene ring results in a signal shift at ∼2.92 ppm. To demonstrate the stability of these Pt NW catalysts, they were recycled via centrifugation between each reaction run (40 cycles). Almost identical performances for the catalyst were observed for each cycle as shown in Fig. 1B. The absolute yield of C went up to 99%. As shown in Fig. 1C and 1D, no obvious differences were observed between the TEM images for the Pt nanocatalysts before the first run and after 40 reaction cycles. This demonstrates the physical stability of the Pt catalysts.
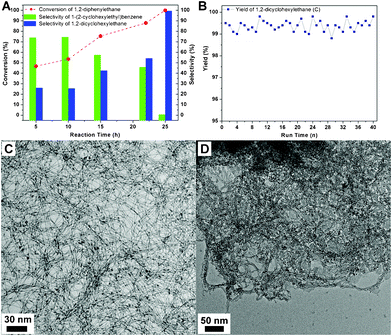 | ||
| Fig. 1 A: Time dependent plots for 1,2-diphenylethane conversion and the selectivity of the hydrogenated products at 70 °C; Pt nanowire (0.005 mmol), 1,2-diphenylethane (1.0 mmol), HOAc (2 mL), AlCl3 (0.25 mmol) and initial hydrogen pressure (1 MPa)). B: Stability tests for the Pt nanocatalyst; typical TEM images of the Pt nanocatalysts: as prepared (C) and after 40 reactions (D). | ||
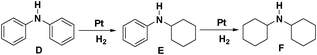
We also investigated the hydrogenation of diphenylamine (D) to further test the catalytic ability of Pt NWs (Table 2). It was interesting to observe that the hydrogenation occurred stepwise which is different from the hydrogenation of 1,2-diphenylethane. Partially reduced intermediate, N-cyclohexylbenzenamine (E) was the only product at the first 15 h in the hydrogenation and the fully hydrogenated compound dicyclohexylamine (F) was obtained only after D converted to E completely. The yield of F was >99% after 30 h reaction time at 70 °C and 1 MPa initial hydrogen pressure in the presence of 0.25 mmol AlCl3. When the initial hydrogen pressure was increased to 2 MPa, 99% of F was successfully obtained within 20 h. In contrast, decreased initial hydrogen pressure (0.5 MPa) reduced the reaction rate. We only obtained 15.9% of F (>99% conversion of D) after 25 h. Furthermore, increasing or decreasing the amount of AlCl3 would affect the catalytic ability of Pt NWs, resulting in low conversion of D.
| Entry | t (h) | p(H2) (Mpa) | Conv. (%) | Select. (%) | |
|---|---|---|---|---|---|
| E | F | ||||
| a Reaction conditions: 0.005 mmol Pt nanowire, 1 mmol diphenylamine, 2 mL HOAc and 0.25 mmol AlCl3 at 70 °C. b 0.125 mmol AlCl3. c 0.5 mmol AlCl3. The conversion and selectivity were determined by GC. | |||||
| 1 | 5 | 1 | 54.9 | 100 | 0 |
| 2 | 10 | 1 | 60.2 | 100 | 0 |
| 3 | 15 | 1 | >99 | 91.6 | 8.4 |
| 4 | 20 | 1 | >99 | 23.7 | 76.3 |
| 5 | 25 | 1 | >99 | 11.5 | 88.5 |
| 6 | 30 | 1 | >99 | <1 | >99 |
| 7b | 25 | 1 | 86.2 | 65.2 | 34.8 |
| 8c | 25 | 1 | 94.2 | 88.6 | 11.4 |
| 9 | 25 | 0.5 | >99 | 84.1 | 15.9 |
| 10 | 20 | 2 | >99 | <1 | >99 |
The general reaction pathway for the hydrogenation of aromatic compounds is shown in Scheme 1. Firstly, one of the benzene molecules adsorbs onto the Pt NW surface, which has been demonstrated by numerous theoretical calculations.14 Then, the absorbed benzene was activated by the Lewis acid (AlCl3).6,13 A sequential step is hydrogenation by hydrogen atoms which were activated by the Pt NWs. These two types of activation work cooperatively, resulting in high activity for producing cyclohexane derivatives.
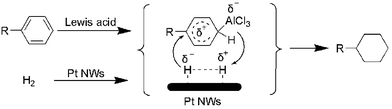 | ||
| Scheme 1 General reaction pathway for the hydrogenation of aromatic compounds. | ||
Another challenge with industrial importance is the hydrogenation of phenol to form cyclohexanone and related cyclohexane derivatives (for nylon 6 and nylon 66).15 We first performed the experiments using Pt NWs as the catalyst in water, formic acid, propionic acid and HOAc (Table 3). No conversions were observed in water or formic acid and little conversion (12.7%) of cyclohexanol (H) was observed in propionic acid. However, the conversion drastically increased to 95.5% in HOAc and cyclohexyl acetate (J) formed from the esterification of HOAc and cyclohexanol (H), was observed as the main product (65.6%). Adding AlCl3 would alter the rate of the reaction. Higher conversion was detected in both water (39.7%) and HOAc (99.6%). The overall selectivity of cyclohexanol and esterified product (H + J) and cyclohexanone (I) was also slightly increased to 72.2(37.6 + 34.6):27.8. This reduction process included three steps: interaction with the Pt NWs surface through the benzene ring; activation of the benzene ring by the Lewis acid and hydrogenation using the activated hydrogen on the surface of the Pt NWs. The benzene ring in phenol is partially hydrogenated to the enol, which can isomerize rapidly to give cyclohexanone. Also, the benzene ring in the phenol can be hydrogenated completely to cyclohexanol and then esterified to cyclohexyl acetate in acetic acid. In general, Pt NWs can be used as an effective catalyst in phenol ring hydrogenation.
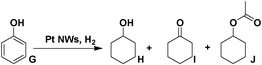
| Entry | Solvent | Cocat. | Conv. (%) | Select. (%) | ||
|---|---|---|---|---|---|---|
| H | I | J | ||||
| a Reaction conditions: 0.005 mmol Pt nanowire, 1 mmol phenol, 2 mL solvent and 0.25 mmol AlCl3 at 60 °C under 2 MPa H2. The conversion and selectivity were determined by GC. | ||||||
| 1 | H2O | — | 0 | 0 | 0 | 0 |
| 2 | HCOOH | — | 0 | 0 | 0 | 0 |
| 3 | C2H5COOH | — | 12.7 | 100 | 0 | 0 |
| 4 | HOAc | — | 95.5 | 0 | 34.4 | 65.6 |
| 5 | H2O | AlCl3 | 39.7 | 12.7 | 87.3 | 0 |
| 6 | HOAc | AlCl3 | 99.8 | 37.6 | 27.8 | 34.6 |
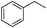
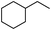
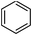
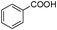
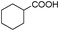
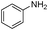
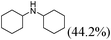
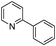
Our Pt NWs were also used as the catalyst in other aromatic compound hydrogenation to illustrate the general applicability as summarized in Table 4. Stilbene and diphenylacetylene were both fully hydrogenated quantitatively to 1,2-dicyclohexylethane in 24 h (Table 4, entries 1 and 2). Ethylcyclohexane was obtained from ethylbenzene hydrogenation with 77.1% conversion after 24 h and quantitatively converted after 48 h (Table 4, entry 3). Benzene can also be reduced to cyclohexane using Pt NWs as the catalyst at 40 °C (Table 4, entry 7) with high yield. Similarly, benzoic acid can be reduced to cyclohexanecarboxylic acid with 53% and >98% yield in 20 and 48 h respectively (Table 3, entry 4). In the case of aniline hydrogenation (Table 4, entry 5), cyclohexanamine (55.8%) and dicyclohexylamine (44.2%) were obtained. We postulate that dicyclohexylamine was obtained due to the deamination of cyclohexanamine. In the case of 2-phenylpyridine hydrogenation, it was interesting to observe selective hydrogenation of benzene over pyridine to yield 2-cyclohexylpyridine almost quantitatively (Table 4, entry 6). It can be explained that the benzene ring can be activated easier than the pyridine ring in this catalytic system which results in the benzene ring hydrogenation with high selectivity.16 This also demonstrated the selectivity of our Pt NW catalytic system.




Conclusions
In conclusion, Pt NWs have been successfully used as highly active and selective catalysts for the direct hydrogenation of various aromatic compounds. The resulting cyclohexane derivatives are both synthetically challenging and industrially important targets. These Pt nanocatalysts are versatile and can be recycled and reused with high conversion efficiency for up to 40 cycles. Mild reaction conditions and low reaction temperatures and initial hydrogen pressures render these catalysts as attractive choices for industrial and synthetic application.Acknowledgements
H.W.G. acknowledges financial support from the National Natural Science Foundation of China (No. 21003092), the Key Project of Chinese Ministry of Education (No. 211064), the Priority Academic Program Development of Jiangsu Higher Education Institutions; X.Q.C. gratefully thanks the financial support from the National Engineering Laboratory for Modern Silk, Soochow University, China; L.H. acknowledges financial support from scientific innovation research of college graduates in Jangsu province (CXZZ11_0102), China. H.-h.Y. thanks the financial support from RIKEN Advanced Science Institute and Grant-in-Aid for Young Scientist (No. 22681016), JSPS/MEXT, Japan.References
- (a) C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987 CrossRef CAS; (b) J. Huang, Y. Jiang, N. Vegten, M. Hunger and A. Baiker, J. Catal., 2011, 281, 352 CrossRef CAS; (c) Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362 CrossRef CAS; (d) P. Makowski, R. D. Cakan, M. Antonietti, F. Goettmann and M.-M. Titirici, Chem. Commun., 2008, 999 RSC.
- (a) C. J. Boxwell, P. J. Dyson, D. J. Ellis and T. Welton, J. Am. Chem. Soc., 2002, 124, 9334 CrossRef CAS; (b) C. Hubert, A. Denicourt-Nowicki, A. Roucoux, D. Landy, B. Leger, G. Crowyn and E. Monflier, Chem. Commun., 2009, 1228 RSC.
- (a) E. Bayram, J. C. Linehan, J. L. Fulton, J. A. S. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Özkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889 CrossRef CAS; (b) N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549 CrossRef CAS; (c) C. Hubert, A. Denicourt-Nowicki, P. Beaunier and A. Roucoux, Green Chem., 2010, 12, 1167 RSC; (d) Y. Motoyama, M. Takasaki, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2009, 11, 5042 CrossRef CAS; (e) T. Maegawa, A. Akashi, K. Yaguchi, Y. Iwasaki, M. Shigetsura, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2009, 15, 6953 CrossRef CAS.
- E. Bayram, M. Zahmakıran, S. Özkar and R. G. Finke, Langmuir, 2010, 26, 12455 CrossRef CAS.
- (a) S. Toppinen, T.-K. Rantakylä, T. Salmi and J. Aittamaa, Ind. Eng. Chem. Res., 1996, 35, 4424 CrossRef CAS; (b) L. Lu, Z. Rong, W. Du, S. Ma and S. Hu, ChemCatChem, 2009, 1, 369 CrossRef CAS.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250 CrossRef CAS.
- (a) M. Zahmakıran, Y. Tonbul and S. Özkar, J. Am. Chem. Soc., 2010, 132, 6541 CrossRef; (b) M. Zahmakıran, Y. Tonbul and S. Özkar, Chem. Commun., 2010, 46, 1228 Search PubMed; (c) M. Zahmakırana, T. Kodaira and S. Özkar, Appl. Catal., B, 2010, 96, 533 CrossRef.
- J. A. Anderson, A. Athawale, F. E. Imrie, F.-M. McKenna, A. McCue, D. Molyneux, K. Power, M. Shand and R. P. K. Wells, J. Catal., 2010, 270, 9 CrossRef CAS.
- C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816 CrossRef CAS.
- B. Hu, K. Ding, T. Wu, X. Zhou, H. Fan, T. Jiang, Q. Wang and B. Han, Chem. Commun., 2010, 46, 8552 RSC.
- (a) M. Li, L. Hu, X. Q. Cao, H. Y. Hong, J. M. Lu and H. W. Gu, Chem.–Eur. J., 2011, 17, 2763 CrossRef CAS; (b) L. Hu, X. Q. Cao, D. H. Ge, H. Y. Hong, Z. Q. Guo, L. Chen, X. H. Sun, J. X. Tang, J. W. Zheng, J. M. Lu and H. W. Gu, Chem.–Eur. J., 2011, 17, 14283 CrossRef CAS.
- Y. Jiang, J. Hess, T. Fox and H. Berke, J. Am. Chem. Soc., 2010, 132, 18233 CrossRef CAS.
- (a) R. R. Deshmukh, J. W. Lee, U. S. Shin, J. Y. Lee and C. E. Song, Angew. Chem., Int. Ed., 2008, 47, 8615 CrossRef CAS; (b) K. Y. Koltunov, G. K. S. Prakash, G. Rasul and G. A. Ohah, Tetrahedron, 2002, 58, 5423 CrossRef CAS; (c) P. Tarakeshwar, J. Y. Lee and K. S. Kim, J. Phys. Chem. A, 1998, 102, 2253 CrossRef CAS.
- (a) F. Delbecq, D. Loffreda and P. Sautet, J. Phys. Chem. Lett., 2010, 1, 323 CrossRef CAS; (b) C. Morin, D. Simon and P. Sautet, Surf. Sci., 2006, 600, 1339 CrossRef CAS; (c) G. Zhong, B. Chan and L. Radom, J. Am. Chem. Soc., 2007, 129, 924 CrossRef CAS; (d) E. Schmidt, A. Vargas, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 12358 CrossRef CAS; (e) A. Valcárcel, A. Clotet, J. M. Ricart, F. Delbecq and P. Sautet, J. Phys. Chem. B, 2005, 109, 14175 CrossRef.
- I. Dodgson, K. Griffin, G. Barberis, F. Pignataro and G. Tauszik, Chem. Ind., 1989, 830 CAS.
- J. C. Cheng, J. Maioriello and J. W. Larsen, Energy Fuels, 1989, 3, 321 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental procedures and the time-dependent 1H NMR analysis of 1,2-diphenylethane hydrogenation. See DOI: 10.1039/c2ra01097f/ |
| This journal is © The Royal Society of Chemistry 2012 |