Solvent-free isomerization of allylic alcohols catalyzed by a rhodium catalyst-organic framework†
Elizabeth G.
Corkum
,
Suneth
Kalapugama
,
Michael J.
Hass
and
Steven H.
Bergens
*
Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2. E-mail: steve.bergens@ualberta.ca; Fax: (780) 493-8231; Tel: (780) 492-9703
First published on 7th February 2012
Abstract
The solvent-free isomerization of allylic alcohols is reported using an immobilized rhodium catalyst-organic framework (COF) synthesized from alternating ring-opening metathesis polymerization (altROMP) assembly. The catalyst provided TONs as high as 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, with catalyst loadings as low as 0.0025 mol% for the isomerizations. Kinetic resolution of starting allylic alcohol 5 was also observed.
000, with catalyst loadings as low as 0.0025 mol% for the isomerizations. Kinetic resolution of starting allylic alcohol 5 was also observed.
We report solvent-free isomerizations of allylic alcohols into the corresponding aldehydes and ketones over a rhodium-diphosphine catalyst that is incorporated into a heterogeneous, polymeric catalyst-organic framework (COF). A prominent strategy to minimize the cost and environmental impact of synthesis is to develop atom-economical reactions that occur over reusable catalysts in inexpensive, non-toxic solvents.1 The catalytic isomerization of allylic alcohols into the corresponding aldehydes or ketones is an ideal candidate for such a transformation because it occurs with 100% atom economy and because it produces useful, versatile products.2 For example, the related rhodium-BINAP (BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) catalyzed enantioselective isomerization of diethylgeranylamine (to form the corresponding aldehyde after hydrolysis) is a fundamental step in the industrial preparation of (−)-menthol.3 Extensive mechanistic studies have been carried out on this type of isomerization that generally conclude that these reactions proceed via an intramolecular net 1,3-hydride shift.2
As is the norm for transition-metal catalyzed reactions, isomerizations of allylic alcohols are mostly carried out in organic media.2,4 The common catalysts for this reaction contain precious metals such as rhodium,4a–c iridium4d–g and ruthenium.4h–q There are examples, however, of catalysts containing non-precious metals such as nickel5a,b and iron.5c–e Ruthenium catalyst systems are the most active to date,4h–q while rhodium- and iridium-containing chiral catalysts are commonly used for enantioselective isomerizations.4a–g These isomerizations likely proceed with the corresponding enols as intermediates. Indeed, an earlier report shows that rhodium-bis(phosphine) catalysts produce the corresponding enols in high concentrations under rigorously neutral conditions.6
Recent studies on low-impact solvents include the use of water,7 ionic liquids,7l and aqueous-biphasic8 mixtures with homogeneous catalysts. There are several reports of reusing the catalyst in biphasic solvent systems. The catalyst loadings per run in these systems range from 0.2 to 1.58 mol%.7d,e,i,k–m,8d Most report 4 reuses, highest number is 9.7d,k The highest total number of turnovers is 2035, obtained after 4 reuses of a ruthenium(IV)-bis(allyl) catalyst.7i To the best of our knowledge, there is only one study reporting the use of a heterogenized catalyst for these isomerizations. A ruthenium-arene-1,3,5-triaza-7-phosphatricyclo [3.3.1.1] decane compound, tethered to Fe3O4 nanoparticles, sustained up to 3 reuses without significant loss in activity. The substrate loadings per run were modest, to result in a total turnover number (TON) of 283, after 4 resues.7m There is only one study of a solvent-free isomerization: the conversion of neat 3-butene-2-ol into 2-butanone with a ruthenium-bis(bipyridine) derivative as catalyst (sub : cat = 1000:1, 80 °C, 10 min) reported by Lau et al.4i There are no studies of solvent-free isomerizations of allylic alcohols over a heterogenized catalyst.
As general strategies, homogeneous catalysts are typically immobilized via non-covalent and covalent interactions with a support. Non-covalent immobilization techniques9 include electrostatic interactions between charged catalysts and supports, physisorption onto the support, hydrogen bonding, and encapsulation within the support. Covalent immobilization techniques10 include the formation of metal-support bonds and formation of bonds between a modified ligand and a support. Grafting of modified ligands onto polymeric supports by copolymerization of modified catalyst ligands is often achieved by first incorporating the ligand into the support followed by metalating the incorporated ligand. This approach often results in incomplete metalation of the ligand-polymer support and poor mass transport at the active sites, leading to low catalyst activity and poor reusability. The direct polymerization of metal-containing monomers ensures metalation of the ligand-polymer support,11 but mass transport at the catalytic active sites must still be addressed.
We reported12 the synthesis of a ruthenium-BINAP-containing COF via an alternating ring-opening metathesis polymerization (altROMP). This ruthenium-based COF was reused for an enantioselective ketone hydrogenation with high TONs per run (> 1000) for 25 runs before loss in activity occurred. More than 35 reuses were carried out without loss in enantioselectivity or detectable leaching of ruthenium. We recently applied the altROMP method13 to prepare a rhodium-BINAP-containing COF (1). COF 1 was synthesized via altROMP between the rhodium(I) chloro-bridged dimer [RhCl(2)]2 (3, 2 = (R)-5,5′-dinorimido-BINAP) and cis-cyclooctene (COE) using the first-generation Grubbs ROMP catalyst, RuCl2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh)(PCy3)2 (4) (Scheme 1). Briefly, the difference in ring strain and crowding between the norbornene groups in 3 and COE results in an alternating ROMP that assembles the three dimensional, cross-linked COF 1. The COF was deposited as a thin film onto BaSO4 to provide mechanical stability and to aid mass transport during catalysis. To our knowledge, this framework is the first polymer-based heterogenized catalyst that operates with higher TONs and selectivity than the parent homogeneous catalyst for a particular reaction.13 Specifically, we reused 1 (after one activation with AgSbF6) up to seven times (TONs = 100 per run) for the highly enantioselective intramolecular cycloisomerization of 1,6-enynes. In batch mode, 1 effected the cycloisomerization of CyC
CHPh)(PCy3)2 (4) (Scheme 1). Briefly, the difference in ring strain and crowding between the norbornene groups in 3 and COE results in an alternating ROMP that assembles the three dimensional, cross-linked COF 1. The COF was deposited as a thin film onto BaSO4 to provide mechanical stability and to aid mass transport during catalysis. To our knowledge, this framework is the first polymer-based heterogenized catalyst that operates with higher TONs and selectivity than the parent homogeneous catalyst for a particular reaction.13 Specifically, we reused 1 (after one activation with AgSbF6) up to seven times (TONs = 100 per run) for the highly enantioselective intramolecular cycloisomerization of 1,6-enynes. In batch mode, 1 effected the cycloisomerization of CyC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2OCH2CHCH(CH2CH3) with TON = 890 in 2-MeTHF solvent. In comparison, the parent catalyst under typical conditions for the homogeneous reaction (5 mol% (COD)RhCl2, 10 mol% BINAP, 20 mol% AgSbF6 in (CH2Cl)2) formed a complex mixture of products with TON = 10. We now report the results from our study of the utilization of 1 for high TON, solvent-free isomerizations of allylic alcohols.
CCH2OCH2CHCH(CH2CH3) with TON = 890 in 2-MeTHF solvent. In comparison, the parent catalyst under typical conditions for the homogeneous reaction (5 mol% (COD)RhCl2, 10 mol% BINAP, 20 mol% AgSbF6 in (CH2Cl)2) formed a complex mixture of products with TON = 10. We now report the results from our study of the utilization of 1 for high TON, solvent-free isomerizations of allylic alcohols.
 | ||
| Scheme 1 Synthesis of rhodium COF 1. | ||
We initially chose the isomerization of 1-propen-3-ol to evaluate 1. With 0.01 mol% Rh, 0.05 mol% AgSbF6 at 70 °C in the absence of solvent, a TON of 6150 was obtained after 45 min to produce a mixture containing propanal (55%), 1-propen-3-ol (28%), and the hemiacetal from propanal and 1-propen-3-ol (17%). The hemiacetal resulted from carrying out the isomerization in the absence of solvent. We found, however, that the use of secondary allylic alcohols avoided the formation of hemiacetals. For example, the isomerization of neat 1-buten-3-ol (5) over 1 (0.025 mol% Rh, 0.125 mol% AgBF4, 70 °C) was complete in 1.25 h to generate 2-butanone as the sole detectable product in ∼4000 TOs.
A factor that will determine the activity of 1 is the extent of activation of the rhodium centers by removal of chloride by the silver salt. Another factor is the coordinating ability of the silver salt anion. Table 1 shows the results from screening a variety of silver salts for the isomerization of neat 5 into 2-butanone
The SbF6− and the BF4− silver salts produce catalysts from 1 with higher initial activities than the OTf− and ClO4−. The catalyst with the BF4− counter ion also effected the isomerization with the highest TON after 48 h (35200, Table 1, entry 2). The effectiveness of these silver salts to act as chloride extractors from 1 likely relates to their solubility in 5 and to their ability to reach the active sites in 1. We point out that abstraction of chloride from the rhodium centers will convert 1 from a neutral framework cross-linked at rhodium to a more open, charged species. Further investigation is required to determine how the nature of the counter ion would influence the structure and reactivity of such a charged framework. For the purpose of this study, AgBF4 was used as the chloride abstractor.
Table 2 shows the results we obtained from the isomerizations of a variety of secondary allylic alcohols (Scheme 2). Substrates 5, 6 and 7, which contain shorter terminal alkyl chains than substrates 8 and 9, were the most active (Table 2, entries 1–5). Catalyst loadings as low as 0.0025 mol% (Table 2, entry 2) resulted in a TON of 35200 for the isomerization of 5. Supports other than BaSO4 were also shown to be effective (Table 2, entry 3). The use of 1 supported on Ba-(L)-tartrate (0.0025 mol% Rh) resulted in a TON of 38000, the highest TON reported for the rhodium-catalyzed isomerization of allylic alcohols. However, possible differences in swellability of the support resulted in the higher initial activity of 1/BaSO4 (TON of 6400 compared to 3600). Although it was necessary to increase the temperature as the terminal alkyl chain in the substrates increased, the catalyst loadings (0.03–0.0025 mol%) were still low relative to the loadings of rhodium catalysts reported in the literature (0.2–5 mol%).4a–c
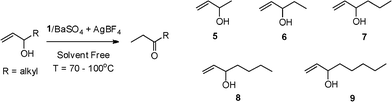 | ||
| Scheme 2 Isomerization of secondary allylic alcohol substrates. | ||
| Entry | Sub | Sub/Ag/Rh | T/°C | Time (h) | TON (TOF, h−1) | Yield (%) |
|---|---|---|---|---|---|---|
| a All runs were carried out under solvent-free conditions with AgBF4 as an activator. b 1/Ba-(L)-Tartrate was the catalyst used. | ||||||
| 1 | 5 | 4000/5/1 | 70 | 1.25 | 3999 | 99 |
| (3199) | ||||||
| 2 | 5 | 40000/5/1 |
70 | 1 | 6400 | 16 |
| (6400) | ||||||
| 2 | 9600 | 24 | ||||
| 24 | 31200 |
78 | ||||
| 48 | 35200 |
88 | ||||
| (733) | ||||||
| 3 | 5 b | 40000/5/1 |
70 | 1 | 3600 | 9 |
| (3600) | ||||||
| 24 | 30000 |
75 | ||||
| 48 | 38000 |
95 | ||||
| (792) | ||||||
| 4 | 6 | 5000/5/1 | 85 | 1.5 | 4999 | 99 |
| (3333) | ||||||
| 5 | 7 | 5000/5/1 | 85 | 1 | 4999 | 99 |
| (4999) | ||||||
| 6 | 8 | 5000/5/1 | 85 | 1.5 | 800 | 16 |
| (533) | ||||||
| 17 | 3150 | 63 | ||||
| 48 | 4500 | 90 | ||||
| (94) | ||||||
| 7 | 9 | 3000/5/1 | 100 | 22 | 2550 | 85 |
| (116) | ||||||
Table 3 shows the results from a comparison of the isomerization of 5 carried out with the homogeneous catalyst system (0.0025 mol% [RhCl((R)-BINAP)]2, 0.0125 mol% AgBF4) under the same solvent-free conditions at 70 °C. It is evident that the immobilized catalyst 1/BaSO4 is almost twice as active as the homogeneous catalyst in the isomerization of 5. To our knowledge, catalyst 1/BaSO4 is the only immobilized polymer-based catalyst that is more active than the parent homogeneous catalyst.
To our knowledge, there are three reports14,15 on the kinetic resolution of allylic alcohols by isomerization (Scheme 3). The study by Noyori14b reports the highest ee (91%) to date, obtained with the kinetic resolution of 4-hydroxy-2-cyclopentenone by a rhodium-BINAP catalyst. This isomerization required 14 days at 0 °C and proceeded in 27% yield. The most recent study by Crochet and Gimeno14c reported the use of [RuCl2(η6-arene){(R)-PR-(binaphthoxy)}]-type catalysts in the (S)-enantio-enrichment of 1-phenyl-2-propen-1-ol and related allylic alcohols with ee's ranging from 4–17% (1 mol% cat., 2 mol% KOtBu, in THF solvent at T = 45–75 °C).
 | ||
| Scheme 3 Kinetic resolution of allylic alcohols. | ||
Using 1/BaSO4 (0.025 mol% Rh, 0.12 mol% AgBF4) at 30 °C in the absence of solvent, we obtained 38% yield of 2-butanone in the isomerization of substrate 5 after 21 h. The remaining 62% of unreacted 5 had an ee of approximately 13%. Studies on the optimization of reaction conditions and choice of support are underway in our laboratories.
In conclusion, we have presented the altROMP assembly of the BaSO4 supported COF 1 and its high activity in the solvent-free isomerization of allylic alcohols. Turnover numbers as high as 38000 were achieved in single-batch reactions, with loadings that are 100–800 times less than is typical for rhodium-based catalysts reported in the literature. We note the highest number of turnovers to date is 1500000, reported for the isomerization of 1-octen-3-ol with an achiral ruthenium(IV)-bis(allyl) catalyst.7i
Our immobilized catalyst also provided higher turnover numbers than the parent homogeneous system, a rare occurrence in the field of catalysis. The possible origins of the COF's activity include support (BaSO4) effects, catalyst-framework interactions, and encapsulation phenomena. Our current research is focused upon optimizing the structure of 1, investigating the origins of its activity, and, in lieu of traditional batch reuses, incorporating 1 into continuous flow reactors.
Acknowledgements
This work was supported by the National Sciences and Engineering Research Council of Canada (NSERC) and the University of Alberta.References
- (a) J. A. Linthorst, Found. Chem., 2010, 12, 55–68 CrossRef CAS; (b) I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef; (c) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998; (d) F. M. Kerton, Alternative Solvents for Green Chemistry, Royal Society of Chemistry, 2009 Search PubMed.
- (a) For recent reviews see: R. C. van der Drift, E. Bouwman and E. Drent, J. Organomet. Chem., 2002, 650, 1–24 CrossRef CAS; (b) R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27–51 CrossRef CAS; (c) L. Mantilli and C. Mazet, Chem. Lett., 2011, 40, 341–344 CrossRef CAS; (d) V. Cadierno, P. Crochet and J. Gimeno, Synlett, 2008, 1105–1124 CAS; (e) G. C. Fu, in Modern Rhodium-Catalyzed Organic Reactions, Wiley-VCH, 2005, 79–91 Search PubMed; (f) A. Varela-Álvarez, J. A. Sordo, E. Piedra, N. Nebra, V. Cadierno and J. Gimeno, Chem.–Eur. J., 2011, 17, 10583–10599 CrossRef.
- (a) K. Tani, T. Yamagata, S. Otsuka, S. Akutagawa, H. Kumobayashi, T. Taketomi, H. Takaya, A. Miyashita and R. Noyori, J. Chem. Soc., Chem. Commun., 1982, 600–601 RSC; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, 1994, Chapter 3 Search PubMed.
- (a) K. Tanaka, S. Qiao, M. Tobisu, M. M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 9870–9871 CrossRef CAS; (b) K. Tanaka and G. C. Fu, J. Org. Chem., 2001, 66, 8177–8186 CrossRef CAS; (c) M. T. Reetz and H. Guo, Synlett, 2006, 2127–2129 CrossRef CAS; (d) L. Mantilli, D. Gérard, S. Torche, C. Besnard and C. Mazet, Chem.–Eur. J., 2010, 16, 12736–12745 CrossRef CAS; (e) L. Mantilli and C. Mazet, Tetrahedron Lett., 2009, 50, 4141–4144 CrossRef CAS; (f) L. Mantilli and C. Mazet, Chem. Commun., 2010, 46, 445–447 RSC; (g) L. Mantilli, D. Gérard, S. Torche, C. Besnard and C. Mazet, Angew. Chem., Int. Ed., 2009, 48, 5143–5147 CrossRef CAS; (h) P. N. Liu, K. D. Ju and C. P. Lau, Adv. Synth. Catal., 2011, 353, 275–280 CrossRef CAS; (i) R. Uma, M. K. Davies, C. Crévisy and R. Grée, Eur. J. Org. Chem., 2001, 3141–3146 CrossRef CAS; (j) B. Martín-Matute, K. Bogár, M. Edin, F. B. Kaynak and J.-E. Bäckvall, Chem.–Eur. J., 2005, 11, 5832–5842 CrossRef; (k) R. C. van der Drift, M. Gagliardo, H. Kooijman, A. L. Spek, E. Bouwman and E. Drent, J. Organomet. Chem., 2005, 690, 1044–1055 CrossRef CAS; (l) M. Ito, S. Kitahara and T. Ikariya, J. Am. Chem. Soc., 2005, 127, 6172–6173 CrossRef CAS; (m) G. Sabitha, S. Nayak, M. Bhikshapathi and J. S. Yadav, Org. Lett., 2011, 13, 382–385 CrossRef CAS; (n) Y. Takai, R. Kitaura, E. Nakatani, T. Onishi and H. Kurosawa, Organometallics, 2005, 24, 4729–4733 CrossRef CAS; (o) P. Crochet, M. A. Fernández-Zúmel, J. Gimeno and M. Scheele, Organometallics, 2006, 25, 4846–4849 CrossRef CAS; (p) M. Batuecas, M. A. Esteruelas, C. García-Yebra and E. Oñate, Organometallics, 2010, 29, 2166–2175 CrossRef CAS; (q) A. Bouziane, B. Carboni, C. Bruneau, F. Carreaux and J.-L. Renaud, Tetrahedron, 2008, 64, 11745–11750 CrossRef CAS.
- (a) H. Bricout, E. Monflier, J.-F. Carpentier and A. Mortreux, Eur. J. Inorg. Chem., 1998, 1739–1744 CrossRef CAS; (b) D. Cuperly, J. Petrignet, C. Crévisy and R. Grée, Chem.–Eur. J., 2006, 12, 3261–3274 CrossRef CAS; (c) V. Branchadell, C. Crévisy and R. Grée, Chem.–Eur. J., 2003, 9, 2062–2067 CrossRef CAS; (d) H. Cherkaoui, M. Soufiaoui and R. Grée, Tetrahedron, 2001, 57, 2379–2383 CrossRef CAS; (e) C. Crévisy, M. Wietrich, V. Le Boulaire, R. Uma and R. Grée, Tetrahedron Lett., 2001, 42, 395–398 CrossRef.
- J. A. Wiles, C. E. Lee, R. McDonald and S. H. Bergens, Organometallics, 1996, 15, 3782–3784 CrossRef CAS.
- (a) N. Ahlsten, H. Lundberg and B. Martín-Matute, Green Chem., 2010, 12, 1628–1633 RSC; (b) D. A. Knight and T. L. Schull, Synth. Commun., 2003, 33, 827–831 CrossRef CAS; (c) D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746–2747 CrossRef CAS; (d) P. Crochet, J. Díez, M. A. Fernández-Zúmel and J. Gimeno, Adv. Synth. Catal., 2006, 348, 93–100 CrossRef CAS; (e) A. E. Díaz-Álvarez, P. Crochet, M. Zablocka, C. Dunhayon, V. Cadierno, J. Gimeno and J. P. Majoral, Adv. Synth. Catal., 2006, 348, 1671–1679 CrossRef; (f) V. Cadierno, S. E. García-Garrido and J. Gimeno, Chem. Commun., 2004, 232–233 RSC; (g) B. Lastra-Barriera, J. Díez and P. Crochet, Green Chem., 2009, 11, 1681–1686 RSC; (h) T. Campos-Malpartida, M. Fekete, F. Joó, Á Kathó, A. Romerosa, M. Saoud and W. Wojtków, J. Organomet. Chem., 2006, 693, 468–474 CrossRef; (i) V. Cadierno, S. E. García-Garrido, J. Gimeno, A. Varela-Álvarez and J. A. Sordo, J. Am. Chem. Soc., 2006, 128, 1360–1370 CrossRef CAS; (j) B. González, P. Lorenzo-Luis, M. Serrano-Ruiz, É. Papp, M. Fekete, K. Csépke, K. Ősz, Á. Kathó, F. Joó and A. Romerosa, J. Mol. Catal. A: Chem., 2010, 326, 15–20 CrossRef; (k) A. Azua, S. Sanz and E. Peris, Organometallics, 2010, 29, 3661–3664 CrossRef CAS; (l) J. García-Álvarez, J. Gimeno and F. J. Suárez, Organometallics, 2011, 30, 2893–2896 CrossRef; (m) S. E. García-Garrido, J. Francos, V. Cadierno, J.-M. Basset and V. Polshettiwar, ChemSusChem, 2011, 4, 104–111 CrossRef; (n) A. Pontes da Costa, J. A. Mata, B. Royo and E. Peris, Organometallics, 2010, 29, 1832–1838 CrossRef CAS.
- (a) C. Bianchini, A. Meli and W. Oberhauser, New J. Chem., 2001, 25, 11–12 RSC; (b) H. Bricout, E. Monflier, J.-F. Carpentier and A. Mortreaux, Eur. J. Inorg. Chem., 1998, 1739–1744 CrossRef CAS; (c) M. Fekete and F. Joó, Catal. Commun., 2006, 7, 783–786 CrossRef CAS; (d) V. Cadierno, P. Crochet, S. E. García-Garrido and J. Gimeno, Dalton Trans., 2004, 3635–3641 RSC.
- (a) For recent reviews see: J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS; (b) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS; (c) P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122 RSC; (d) X. S. Zhao, X. Y. Bao, W. Guo and F. Y. Lee, Mater. Today, 2006, 9(3), 32–39 CrossRef CAS.
- (a) For recent reviews see: B. M. L. Dioos, I. F. J. Vankelecom and P. A. Jacobs, Adv. Synth. Catal., 2006, 348, 1413–1446 CrossRef CAS; (b) N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS; (c) Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3466 CrossRef CAS; (d) Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322–359 CrossRef CAS; (e) K. Ding, Z. Wang, X. Wang, Y. Liang and X. Wang, Chem.–Eur. J., 2006, 12, 5188–5197 CrossRef CAS; (f) For MOFs see: Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4135 CrossRef CAS; (g) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS; (h) L. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- P. Mastrorilli and C. F. Nobile, Coord. Chem. Rev., 2004, 248, 377–395 CrossRef CAS.
- (a) C. K. Ralph, O. M. Akotsi and S. H. Bergens, Organometallics, 2004, 23, 1484–1486 CrossRef CAS; (b) C. K. Ralph and S. H. Bergens, Organometallics, 2007, 26, 1571–1574 CrossRef CAS.
- E. G. Corkum, M. J. Hass, A. D. Sullivan and S. H. Bergens, Org. Lett., 2011, 13, 3522–3525 CrossRef CAS.
- (a) K. Ohkubo, T. Ohgushi, T. Kusaga and K. Yoshinaga, Inorg. Nucl. Chem. Lett., 1977, 13, 631–636 CrossRef CAS; (b) M. Kitamura, K. Manabe and R. Noyori, Tetrahedron Lett., 1987, 28, 4719–4720 CrossRef CAS; (c) M. A. Fernández-Zúmel, B. Lastra-Barriera, M. Scheele, J. Díez, P. Crochet and J. Gimeno, Dalton Trans., 2010, 39, 7780–7785 RSC.
- Bergens et al. reported the kinetic resolution of 3-buten-2-ol, however the enol, rather than the ketone, product was obtained. For more information see ref. 6.
Footnote |
| † Electronic Supplementary Information (ESI) available: Text giving experimental procedures for catalyst-organic framework preparation and use and kinetic resolution determination. See DOI: 10.1039/c2ra20197f/ |
| This journal is © The Royal Society of Chemistry 2012 |