An efficient oxa-Michael addition to diethyl vinylphosphonate under mild reaction conditions†‡
Ondřej
Baszczyňski
,
Petr
Jansa
,
Martin
Dračínský
,
Martin Maxmilian
Kaiser
,
Petr
Špaček
and
Zlatko
Janeba
*
Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i., Flemingovo nám. 2, 16610, Prague 6, Czech Republic. E-mail: janeba@uochb.cas.cz
First published on 15th December 2011
Abstract
An oxa-Michael addition of various secondary and branched primary alcohols to diethyl vinylphosphonate was systematically studied and optimized. This efficient method precedes using of harsh reaction conditions (e.g. strong bases, high temperatures) and gains access to an important class of biologically active compounds in one step.
Acyclic nucleoside phosphonates (ANPs)1 represent catabolically stable nucleotide analogues with a wide variety of biological properties.2 Although ANPs bearing an unbranched phosphono-ethoxyethyl (PEE) moiety3 usually lack any antiviral activity, 6-oxopurine PEE derivatives have been found to display very promising antimalarial activity.4 To further study structure–activity relationship of ANPs, an efficient synthetic method for the introduction of the phosphonoethyl motif to various secondary alcohols is urgently needed.
Only few successful methods on how to introduce the phosphonoethyl moiety to alcohols have been reported, but with limited applicability and/or poor yields. Reactions of bromoethyl-phosphonate (and the corresponding tosyl or mesyl analogues) with secondary alcohol of 1,2-di-O-tert-butyldimethylsilyl-L-threose did not afford the expected alkylation product, possibly due to β-elimination of the ethylphosphonate derivatives under the basic conditions.5 An introduction of the hydroxyethyl group, followed by the subsequent reaction with trialkylphosphite under Arbuzov reaction conditions afforded dialkyl ethylphosphonate derivatives in low overall yields under harsh reaction conditions.5,6 Finally, 2-hydroxyethylphosphonates were directly introduced to a phenolic hydroxyl under the Mitsunobu reaction conditions, although in 26–54% yields only.7
Thus, the most promising method for the attachment of the ethylphosphonate group is addition reaction to dialkyl vinylphosphonate (VP). Recently, aza-Michael8 and sulfa-Michael9 addition to VP derivatives has become a popular synthetic way for preparation of various biologically active molecules, since amines and thiols are usually very good nucleophiles. On the other hand, VP has been neglected as an acceptor in oxa-Michael addition reactions due to the reversibility of the reaction and low reactivity of alcohols, requiring harsh conditions and thus limiting the synthetic scope of the reaction.
An addition of primary alcohols to dialkyl VP has been reported using strong bases10 or carbonates at elevated temperature.11 Similarly, the oxa-Michael additions of secondary alcohols to dialkyl VP (or vinyl phosphinates) were performed with strong bases at elevated temperatures and with moderate yields only.12
Herein we report on the efficient introduction of the ethylphosphonate moiety via optimized oxa-Michael addition of structurally diverse secondary alcohols to diethyl VP.13
First of all, an extensive study of the reaction of fluorohydrine 1 with diisopropyl bromo- and chloroethylphosphonates (2 and 3, Scheme 1) with various bases (LiH, NaH, t-BuOK, K2CO3, Cs2CO3, DBU, NaOH, BuLi, TBAF, Et3N, MeONa, 1.0–1.5 eq.), in various solvents (DMF, ether, dioxane, toluene), and at various reaction temperatures verified that haloethylphosphonates14 are not suitable reagents for O-alkylation reactions.5 In all cases, an evident formation of the side products 5 and 6 was observed, affording the desired product 4 in 10% yield at best.
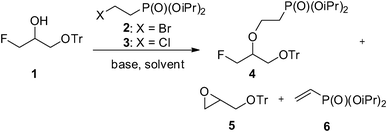 | ||
| Scheme 1 Alkylation of 1 with haloethylphosphonates 2 and 3. | ||
Next, the reaction of fluorohydrine 1 with commercially available diethyl vinylphosphonate (7, DEVP, Scheme 2) under various reaction conditions was studied and thoroughly optimized. The best result (52% of the desired product 8) was observed under the catalysis with KOH in dioxane (entry 5, Table 1).
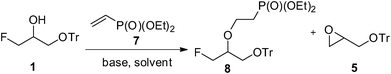 | ||
| Scheme 2 General addition of fluorohydrine 1 to DEVP 7. | ||
| Entry | Base (eq.) | t (°C) | Solvent | Yield of 8 [%]a |
|---|---|---|---|---|
| a Yield (GC-MS). Reaction conditions: DEVP (1.5 eq.), 24 h. | ||||
| 1 | Cs2CO3 (1.0) | rt to 50 | dioxane | 22 |
| 2 | BuLi (0.2) | −70 to rt | THF | 13 |
| 3 | NaH (0.2) | −70 to rt | THF | 33 |
| 4 | NaOH (0.2) | rt | dioxane | 20 |
| 5 | KOH (0.2) | rt | dioxane | 52 |
| 6 | RbOH (0.2) | rt | dioxane | 31 |
| 7 | CsOH (0.2) | rt | dioxane | 34 |
| 8 | t-BuOK (0.2) | rt | dioxane | 39 |
| 9 | DBU (0.1) | rt to 50 | dioxane | 0 |
| 10 | Ba(OH)2 (0.2) | rt to 50 | dioxane | 0 |
The increasing size of the cation had a positive effect on the yields of product 8 (Table 2, Scheme 2), as the reactions with t-BuOK and CsOH reached ~75% conversion in 24 h (with t-BuOLi or t-BuONa less than 5%). A prolonged treatment only led to the increased rate of the retro-Michael reaction (GC-MS).
Sekine et al.15 have described a reaction of the ribonucleoside derivatives with acrylonitrile in t-BuOH in the presence of Cs2CO3 to give 2′-O-cyanoethylated ribonucleosides. Later, their methodology was extended to acrylate esters.16 Although non-activated DEVP (7) is much poorer electrophile compared to acrylates, we decided to apply the Sekine's reaction conditions (1 eq. of Cs2CO3 and 20 eq. of DEVP in t-BuOH) for the addition of fluorohydrine 1 to DEVP (7) and, surprisingly, a quantitative conversion to phosphonate 8 was observed (Scheme 2). The finding, that DEVP (7) can serve as a good Michael acceptor for secondary alcohols under mild reaction conditions, encouraged us to further optimize this reaction.
To reduce amount of the reagents, a set of experiments with gradually decreasing amount of DEVP (7) was carried out (Table 3, Scheme 2). The reaction was completed in 10 h when 5 eq. of starting DEVP (7) were used, in 24 h with only 2 eq. of DEVP, and in prolonged reaction time (48 h) only 1 eq. of DEVP (7) suffices to obtain a 96% conversion to the product 8 (Table 3).
| Time [h] | Yield of 8 [%]a | ||||
|---|---|---|---|---|---|
| 1 eq. | 2 eq. | 5 eq. | 10 eq. | 20 eq. | |
| a Yield determined by GC-MS. Reaction conditions: DEVP (1.0 eq.), Cs2CO3 (1.0 eq.), t-BuOH, rt. | |||||
| 1 | 0.2 | 0.0 | 1.5 | 3.0 | 2.2 |
| 2.5 | 1.3 | 3.5 | 13.3 | 24.2 | 16.6 |
| 5 | 7.1 | 27.8 | 69.4 | 89.5 | 71.0 |
| 10 | 27.0 | 76.2 | 98.8 | 99.4 | 96.5 |
| 24 | 81.4 | 98.3 | 99.5 | 99.9 | 99.9 |
| 48 | 96.0 | 98.6 | 99.5 | 99.9 | 99.9 |
The concentration profile of DEVP (7) in the studied oxa-Michael reaction follows the sigmoid curve typical for autocatalytic reactions (Fig. 1).17 Since the studied reaction (Scheme 2) is heterogeneous due to low solubility of Cs2CO3 in t-BuOH, we speculate that the reaction profile is a result of the solvation effects as phosphonates are known to strongly bind metal ions. Thus, binding of the cesium ion by phosphonate 8 formed during the reaction would increase the solubility of Cs2CO3 and subsequently the reaction rate.
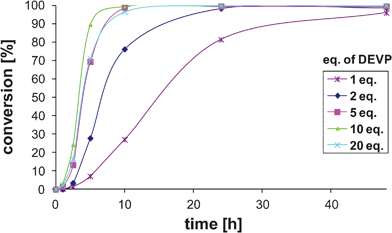 | ||
| Fig. 1 Reaction profile of the oxa-Michal reaction of fluorohydrine 1 as a function of the DEVP (7) concentration. | ||
On the other hand, the catalysis of the studied oxa-Michael addition (Scheme 2) with K2CO3 turned out to be completely ineffective due to even worse solubility of K2CO3 in t-BuOH compared to Cs2CO3. Not even the addition of 1 eq. of [18]crown-6 to the reaction mixture really improved the yield of product 8 (8%, entry 1, Table 4) and the reaction mixture contained mainly the starting compound 1 and epoxide 5 (Scheme 2). Actually, in all experiments using crown ethers (entries 1–4, Table 4), the presence of the “naked” carbonate anion resulted in the preferential formation of epoxide 5. Catalysis of the reaction with KF on alumina (entry 5, Table 4) afforded compound 8 in a satisfactory 68% yield but the results are inferior to the above optimized methodology using Cs2CO3 in t-BuOH. All other experiments with various more or less soluble cesium salts, as well as attempts to reduce the amount of Cs2CO3, resulted in lack of reactivity or extension of the reaction times.
| Entry | Base | Additive | Yields (%)a | ||
|---|---|---|---|---|---|
| 1 | 8 | 5 | |||
| a Yield determined by GC-MS. Reaction conditions: DEVP (1.5 eq.), base (1.0 eq.), crown ether (1.0 eq.), t-BuOH, rt, 24 h. | |||||
| 1 | K2CO3 | [18]crown-6 | 60 | 8 | 32 |
| 2 | Cs2CO3 | [18]crown-6 | 9 | 32 | 59 |
| 3 | Cs2CO3 | DB[21]crown-7 | 2 | 33 | 65 |
| 4 | Cs2CO3 | DB[24]crown-8 | 3 | 52 | 45 |
| 5 | KF/Al2O3 | none | 13 | 68 | 19 |
| 6 | KHDMS | none | 28 | 14 | 58 |
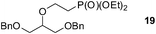
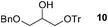
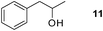
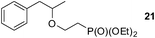
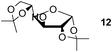
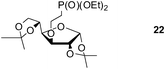
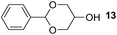
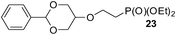
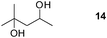
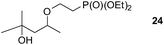
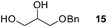
Finally, our optimized reaction conditions of the oxa-Michael addition (alcohol, 1.5 eq. of DEVP, 1 eq. of Cs2CO3, t-BuOH, rt) were subsequently applied to various compounds containing secondary or primary hydroxyl groups (Table 5). Thus, reaction of symmetrically protected glycerol 9 gave high yield of phosphonate 19 (86%, entry 1), while its more sterically hindered derivative 10 afforded phosphonate 20 in lower, but still satisfactory yield (55%, entry 2, Table 5). Structurally diverse secondary alcohols 11–13 were converted to corresponding phosphonates 21–23 in moderate to high yields (51–88%, entries 3–5, Table 5). Selective alkylation of secondary hydroxyl next to tertiary hydroxyl group in compound 14 led to the formation of compound 24 (entry 6), while both primary and secondary hydroxyls of the vicinal diol 15 react to give bisphosphonate 25 (entry 7, Table 5).
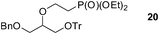
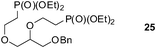
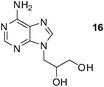
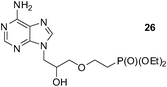
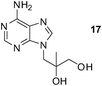
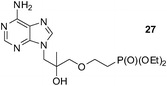
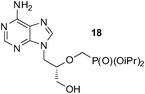
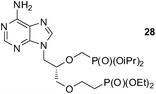
An analogous treatment of acyclic nucleosides 16 and 17 and ANP 18 afforded the desired phosphonoethylated products 26–28 in 12–14% yields only (entries 8–10, Table 5), due to the low solubility of the starting compounds in t-BuOH and presence of the reactive amino group in the C–6 position of the purine ring. To improve synthesis of such potentially biologically active compounds, protection of the amino group with a suitable lipophilic protecting group is strongly advisable.
In summary, the first efficient oxa-Michael reaction of secondary and sterically hindered primary alcohols with diethyl vinylphosphonate (7) under very mild reaction conditions is reported. To demonstrate the importance of our optimized one-step protocol in the field of medicinal chemistry, it is currently being employed for the preparation of wide range of compounds of biological interest.
This study was performed as a part of research project OZ40550506 of the IOCB, v.v.i. and was supported by Ministry of the Interior of the Czech Republic (VG20102015046) and by Gilead Sciences.
References
- E. De Clercq, A. Holý, I. Rosenberg, T. Sakuma, J. Balzarini and P. C. Maudgal, Nature, 1986, 323, 464–467 CrossRef CAS
.
-
(a) A. Holý, Curr. Pharm. Des., 2003, 9, 2567–2592 CrossRef
- A. Holý, I. Rosenberg and H. Dvořáková, Collect. Czech. Chem. Commun., 1990, 55, 809–818 CrossRef
-
(a) D. T. Keough, D. Hocková, A. Holý, L. Naesens, T. S. Skinner-Adams, J. de Jersey and L. W. Guddat, J. Med. Chem., 2009, 52, 4391–4399 CrossRef CAS
- Q. Huang and P. Herdewijn, Nucleosides, Nucleotides Nucleic Acids, 2009, 28, 337–351 CAS
-
(a) G. D. Diana, E. S. Zalay, U. J. Salvador, F. Pancic and B. Steinberg, J. Med. Chem., 1984, 27, 691–694 CrossRef CAS
-
Int. Pat. WO2005/117904 A2, 2005. Search PubMed
-
(a) G. David, B. Boutevin and Y. Hervaud, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 2201–2209 CrossRef CAS
- F. Gao, X. Yan and K. Auclair, Chem.–Eur. J., 2009, 15, 2064–2070 CrossRef CAS
-
(a) H.-J. Kleiner and C. Schumann, Phosphorus and Sulfur, 1982, 13, 363–370 CAS
-
(a) E. Brunet, M. J. Mata, O. Juanes, H. M. H. Alhendawi, C. Cerro and J. C. Rodríguez-Ubis, Tetrahedron: Asymmetry, 2006, 17, 347–354 CrossRef CAS
-
(a) A. N. Pudovik and R. G. Kuzovleva, J. Gen. Chem. USSR, 1963, 33, 2755–2760 CAS
- O. Baszczyňski, P. Jansa, D. Hocková, Z. Janeba, M. Dračínský, A. Holý, D. T. Keough, J. de Jersey and L. W. Guddat, Collect. Symp. Ser., 2011, 12, 302–303 Search PubMed
- P. Jansa, A. Holý, M. Dračínský, O. Baszczyňski, M. Česnek and Z. Janeba, Green Chem., 2011, 13, 882–888 RSC
- H. Saneyoshi, K. Seio and M. Sekine, J. Org. Chem., 2005, 70, 10453–10460 CrossRef CAS
- T. Yamada, N. Okaniwa, H. Saneyoshi, A. Ohkubo, K. Seio, T. Nagata, Y. Aoki, S. Takeda and M. Sekine, J. Org. Chem., 2011, 76, 3042–3053 CrossRef CAS
- R. Plasson, A. Brandenburg, L. Jullien and H. Bersini, J. Phys. Chem. A, 2011, 115, 8073–8085 CrossRef CAS
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra00938b/ |
| ‡ This work is dedicated to Prof. Antonín Holý on the occasion of his 75th birthday. |
| This journal is © The Royal Society of Chemistry 2012 |