Organofunctionalized magnesium phyllosilicates as mono- or bifunctitonal entities for industrial dyes removal
Andréa S. O.
Moscofian
,
Cléo T. G. V. M. T.
Pires
,
Adriana P.
Vieira
and
Claudio
Airoldi
*
Institute of Chemistry, University of Campinas, UNICAMP, P.O. Box 6154, 13084-971, Campinas, SP, Brazil. E-mail: airoldi@iqm.unicamp.br
First published on 2nd March 2012
Abstract
Magnesium phyllosilicates with organic groups anchored onto the inorganic polymeric backbones as mono- or bifunctional entities were investigated for dye removal from aqueous solution. The synthetic methodology consisted in obtaining organofunctionalized nanostructured materials from silylating agents of general formula R1–Si(OCH3)3, in which R1-trimethoxysilane includes chains containing the functional groups: 3-aminopropyl, octadecyldimethyl(silylpropyl)ammonium, 3-mercaptopropyl, 3-ethylenediamine and 3-diethylenetriamine. The sol–gel process leads to lamellar structures similar to those of natural silicate with basal distances, in good agreement with the R1 contained in the precursor agent. The pendant electrophile attached on the new phyllosilicates interacts with the negative charge of dyes used in the textile industry, such as Reactive Yellow GR, Reactive Red RB and Reactive Blue RN. The sorption studies showed that the phyllosilicate containing octadecyldimethyl(3-trimethoxysilylpropyl)ammonium chloride agent, P-OCT, presented the highest sorption capacities of 1343, 1286 and 344 mg g−1 for the dyes Yellow GR, Blue RN and Red RB, respectively, which are better than for other sorbing materials. Real samples from textile effluents assayed demonstrated that the sorption did not need to adjust the initial pH, with surface saturation after 3 h and the minimum mass necessary was 2.5 g dm−3 of P-OCT for the best efficiency. This is thus a very promising material for textile effluent treatment, with good structural disposition of the pendant groups.
Introduction
Environmental problems have become increasingly critical and frequent, due to population growth and increased industrial activity associated with contamination of natural waters which is one of the major problems of modern society. Textile industries contribute to this problem, generating large volumes of effluents that, when left untreated, cause a serious environmental damage.1 However, such kind of industry plays an important role in the economy of many countries, but consumes huge amounts of water, due to the inputs of dyes, detergents, softeners, etc. Thus, large volumes of waste containing high organic loads and strong staining components intensively originate from coloring clothes, paper, houses, cars etc.2The colored dye effluents employed in the fiber dyeing produce high concentrations associated with considerable loads of the effluents that make water treatment difficult.1,2 In general, it is estimated that near 20% of the initial dyes is lost to waste, which represents one of the major environmental problems faced by textile industry.3 When not properly treated, the dye is released into natural waters and effluents can potentially damage the ecosystem, reducing water clarity and blocking solar radiation, which affects the photosynthetic activity and the rate of gaseous solubility.1,4 Another dangerous problem involving dyes is exemplified with azo compounds that cause carcinogenic and mutagenic effects.3,5 In attempting to remove color from effluents, several methods can be applied, such as membrane filtration related to nanofiltration, reverse osmosis, electrodialysis, ion exchange and sorption techniques.5,6
Among these methods, sorption is the most popular and efficient for effluent removal, permitting water reuse, which decreases costs, flexibility, simplicity of design, ease of operation etc. For this methodology there are no problems involved with intermediate formation, depending only on such physical and chemical features as sorbent–dye interaction, sorbent surface area, particle size, temperature, pH and contact time.6
Activated carbon is the most widely used sorbent for dye removal due to its high sorption capacity and efficiency,7,8 as demonstrated for both cationic and acidic dyes. However, this sorbent has several disadvantages, such as high cost and inefficiency with direct and reactive dyes. Due to these drawbacks found with activated carbon, researchers have intensified efforts to produce alternative sorbents to replace it in recent years. Attention has been directed to many alternative solid sorbents that are capable of pollutant removal from contaminated water, such as fly ash, pine bark, chitosan, babassu coconut, bentonite, chemically modified silicas,9–15 among others.
Within a series of synthetic inorganic materials used for dye removal, clay minerals and organoclays act as favorable agents due to the swellability, which increases the facility to embrace dye of various sizes and enable the accommodation of high amounts of them, independent of the pore size or the external surface.16 Thus, depending on the covalent pendant chain bonded to the layer surface, containing available organic groups attached, large amounts of dyes can be sorbed on well-structural organized inorganic–organic layered hybrids.17,18 Also different applications have previously been reported for such class of materials for heavy metal removal,19–23 other pollutants adsorption,24,25 polymer fillers,26 environmental barriers, catalytic supports, improvement of optical and mechanical properties.27,28 It is interesting to notice that also complexes dye–clay have further applications too.29
Based on the search to explore new sorbents, the present investigation deals with organically modified magnesium phyllosilicates, as new agents for dyes removal with high performance, when applied with aqueous industrial textile effluents
2. Results and discussion
The synthetic procedure leads to lamellar layers of the magnesium phyllosilicates formed through the sol–gel process, using generalized trialkoxysilanes, (RO)3Si–R1, where R1 represents the attached organic moieties, as shown in Fig. 1. These agents were hydrolyzed in alkaline conditions, yielding silanol groups to perform the condensation reaction with magnesium hydroxide, to constitute the inorganic Si–O–Mg framework.30 The series of synthesized phyllosilicates is constituted by two monofunctional ones, one neutral with short pendant chains, 3-aminopropyltriethoxysilane (P-AT), and the other with long charged pendant chains, octadecyldimethyl(3-trimethoxysilylpropyl)ammonium chloride (P-OCT). The bifunctional phyllosilicates were synthesized through the equimolar combination of two silylating agents, 3-aminopropyltriethoxysilane and 3-mercaptopropyltrimethoxysilane (P-ATM), 3-diethylenetriaminopropyltrimethoxysilane and 3-mercaptopropyltrimethoxysilane (P-EM), and 3-diethylenetriaminopropyltrimethoxysilane and 3-mercaptopropyltrimethoxysilane (P-DM). The elemental results for the synthesized compounds are listed in Table 1. These five new synthesized phyllosilicates were used to dyes removal, as shown in Fig. 2. However, the best sorption capacity was obtained with P-ATM and P-OCT.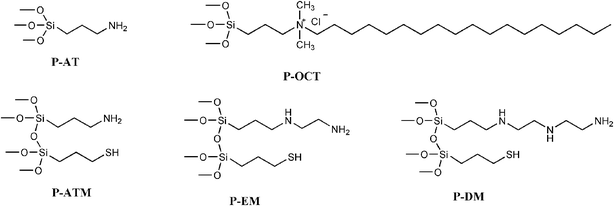 | ||
| Fig. 1 Structure of pendant silylating agents on organically modified mono- and bifunctional magnesium phyllosilicates. | ||
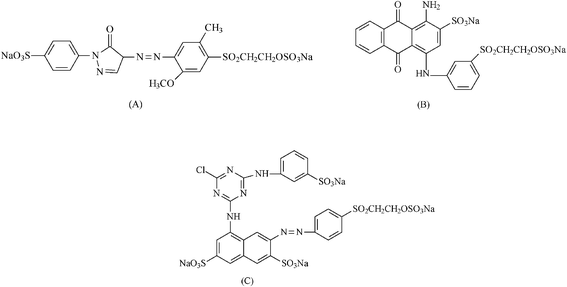 | ||
| Fig. 2 Structure of reactive dyes: Yellow RY (A), Red RB (B) and Blue RN (C). | ||
| Phyl | C/% | H/% | N/% | S/% | Cl/% | ρ/mmol g−1 |
|---|---|---|---|---|---|---|
| P-AT | 16.06 | 4.88 | 7.19 | — | — | 5.13 |
| P-OCT | 47.59 | 9.29 | 3.93 | — | 3.16 | 2.81 |
| P-ATM | 19.44 | 4.94 | 2.95 | 10.34 | — | 5.33 |
| P-EM | 20.98 | 5.89 | 4.96 | 7.34 | — | 4.06 |
| P-DM | 24.59 | 5.97 | 6.74 | 6.76 | — | 3.71 |
The amounts of immobilized pendant groups decrease with increasing the length of the organic chains of the silylating agents in both mono- or bifunctionalized phyllosilicates. For P-AT, which incorporates only simple propyl carbon chains, a reasonable degree of 5.13 mmol g−1 was attached. When pendant propyl chains combined thiol and amino functions, the highest value of 5.33 mmol g−1 for P-ATM was determined. However, the combination formed by the propyl thiol group with addition of an ethylene moiety in the precursor agent, gave a value of 4.06 mmol g−1 for the P-EM compound with the same thiol propyl moiety and longer chains containing three basic nitrogen centers, formed by adding two ethyl groups to the precursor amine chain, synthesizing P-DM, the degree of functionalization decreases to 3.71 mmol g−1. Based on these new synthetic phyllosilicates, it is clearly shown that, as the amine chains increase in number of carbon atoms to obtain the organic chain attached in combination with the fixed thiol agent, the organofunctionalization is proportionally decreased. Finally, the phyllosilicate P-OCT that presents, in addition to propyl chains, an octadecyl chain, showed the smallest functionalization degree of 2.81 mmol g−1. This fact could be explained by steric effects promoted by longer carbon chains, which hinders the process of condensation of the silylating agent to form the hybrid inorganic phase. These results confirm that the degree of functionalization is inversely proportional to the size of the carbon chain of the silylating agent used in the synthetic procedure.
The infrared spectra for the organofunctionalized synthesized magnesium phyllosilicates are shown in Fig. 3, in which the main bands are marked. The broad band centered at 3400 cm−1 is attributed to OH stretching vibrations. Moreover, this band is attributed to hydroxyl group stretching of water molecules that are sorbed on the surface through hydrogen bonds.31 The bands near 2900 cm−1 are due to symmetric and asymmetric CH bond stretching of the carbon chain of each silylating agent. The angular deformation of water molecules sorbed corresponds to the band at 1630 cm−1, that is more evident in the spectrum of Fig. 3b. In other cases, it is followed by bands of the nitrogen–hydrogen deformations that appear at 1570 cm−1 for the phyllosilicates containing amino groups. The band centered at 1200 cm−1 refers to Si–C stretching vibrations.32 The intense bands observed at 1120 and 1050 cm−1 are assigned to the stretching of siloxane bonds, Si–O–Si, which are responsible for the formation of the inorganic phyllosilicate framework,32 and the band at 550 cm−1 is related to the Mg–O inorganic structure deformation.33 For the spectra in Fig. 3a and 3b, there are intense and narrow bands in the 1380 cm−1 region, which are attributed to the presence of nitrate groups, from the original salt used for the phyllosilicte synthesis that are interspersed between the layers of the structures and were not completely removed during the washing process.34,35 On the other hand, this band is absent from the spectra in Fig. 3c to 3e.
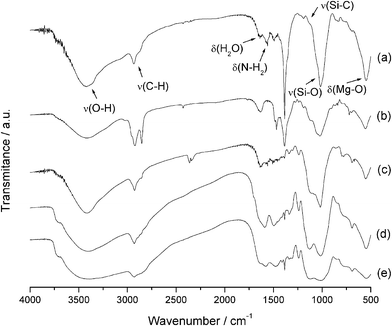 | ||
| Fig. 3 Infrared spectra of P-AT (a), P-OCT (b), P-ATM (c), P-EM (d), P-DM (e) phyllosilicates. | ||
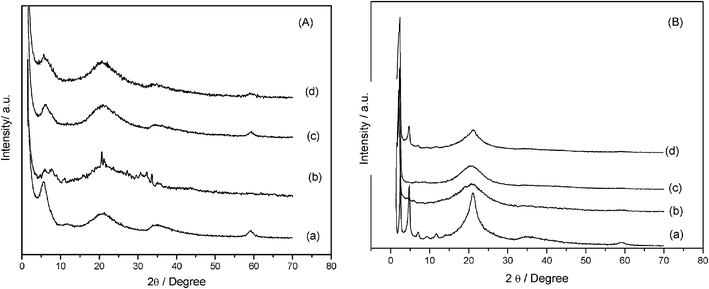
The X-ray diffraction patterns obtained for the phyllosilicates are shown in Fig. 4, indexed according to the basal planes on the structure of talc. The appearance of four well-defined peaks is observed expected from the organofunctionalized phyllosilicates with the known 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trioctahedral structure.36 The first peak between 0 to 10° region represents the (001) crystallographic plane, which corresponds to the basal distance, defined by the sum of interlamellar spacing and the thick layer. Another important peak for phyllosilicate structures is related to the (060) plane, which appears at 2θ values equal to 60°. The presence of this plane indicates that the synthesized phyllosilicates have trioctahedral structures similar to those found in natural talc.37
:1 trioctahedral structure.36 The first peak between 0 to 10° region represents the (001) crystallographic plane, which corresponds to the basal distance, defined by the sum of interlamellar spacing and the thick layer. Another important peak for phyllosilicate structures is related to the (060) plane, which appears at 2θ values equal to 60°. The presence of this plane indicates that the synthesized phyllosilicates have trioctahedral structures similar to those found in natural talc.37
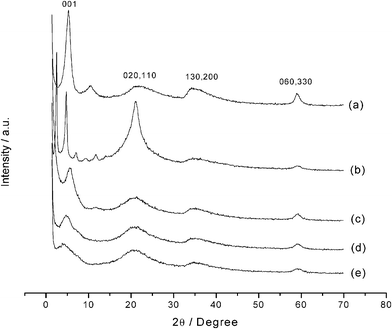 | ||
| Fig. 4 X-Ray diffraction patterns for P-AT (a), P-OCT (b), P-ATM (c), P-EM (d) and P-DM (e) phyllosilicates. | ||
The reflection (020), at 2θ near to 20°, is also observed and has relevance for this type of material for inferring features related to organo-minerals.38 Unlike highly crystalline structures with well-formed crystals, the diffraction peaks of organofunctionalized phyllosilicates present broader reflections, due to the smaller particle sizes, the structural X-ray scattering by the organic groups and also some disorder caused by the insertion of the organic molecules between the lamella or even on the surface.
The peaks associated with P-OCT phyllosilicate are shown in Fig. 4b, with a more intense reflection (020) for 2θ near to 20°, with a narrow shape in comparison to other compounds. This behavior suggests that, for this material, which has a long carbon chain, there is the presence of an organized organic chain in the solid phase39 and, consequently, this phyllosilicate has higher crystallinity than the others.
From the basal angle, related to the (001) plane, the basal distance (d) was calculated by using Bragg's law and the results are listed in Table 2. This value is defined as the distance between the upper ends of two consecutive lamellas. This basal distance of the natural lamellar compound correlated to talc, which has a similar structure to that presented in this investigation, corresponding to a value near to 1.0 nm.40 Looking at the values for all the synthesized structures, the d values are higher than natural talc, in good agreement with increasing in the value with organic groups of different lengths, considering the attached pendant chains in a linear and not bended disposition.
| Phyl | 2θ/° | d 001/pm |
|---|---|---|
| P-AT | 5.3 | 1666 |
| P-OCT | 2.5 | 3531 |
| P-ATM | 5.6 | 1577 |
| P-EM | 4.8 | 1839 |
| P-DM | 4.0 | 2207 |
Silylating agent immobilization into the inorganic phyllosilicate structures can be confirmed by nuclear magnetic resonance of the silicon nucleus in the solid state, through the appearance of Tn peaks in the spectra,41 referring to the R–Si–(O–Si)3, R–Si–(O–Si)2–(R'), with R' = –OH or –OCH3 and R–Si–(O–Si)–(R')2 arrangements, referring to T3, T2 and T1 species, respectively. The spectra obtained for the magnesium phyllosilicates are shown in Fig. 5. There are three species at −50, −56 and −68 ppm, respectively, for all phyllosillicates, with the exception of P-AT, shown in Fig. 5a, that has no T3 species. The appearance of these peaks indicates the existence of the organic chains covalently linked to silicon atom through a silicon–carbon bond, proving the effectiveness of the functionalization process.33
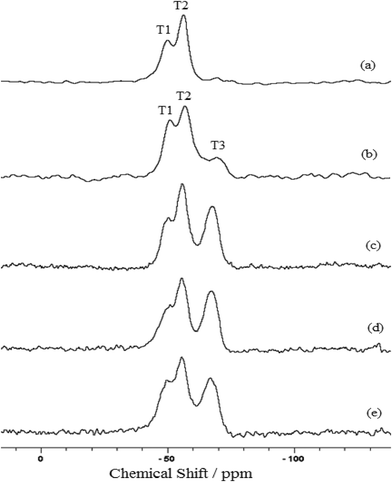 | ||
| Fig. 5 29Si NMR spectras for the P-AT (a), P-OCT (b), P-ATM (c), P-EM (d) and P-DM (e) phyllosilicates. | ||
The talc-like phyllosilicates in powder form, as synthesized in the present investigation, do not show any Q species in their structures, characterized by an absence of peaks that should appear between the −110 and −90 ppm region in the spectra. This fact can be related by the absence of free silanol groups bonded to the inorganic silicon network. The organic pendant groups proceeding from the silylating agents, are the only silicon source used in the synthetic procedures.42
The nuclear magnetic resonance of the carbon nucleus in the solid state for all phyllosilicates was also explored and the spectra obtained are illustrated in Fig. 6. The peaks were assigned according to the numbers marked in the corresponding structures of each compound inserted in the spectra. In the case of P-OCT, the assignment of the signals is based on a previous investigation performed to obtain the same compound, but with hydrothermal treatment.41 Some carbons are unassigned due to the difficulty in localizing the expected chemical shifts for the pendant chains in the spectrum.
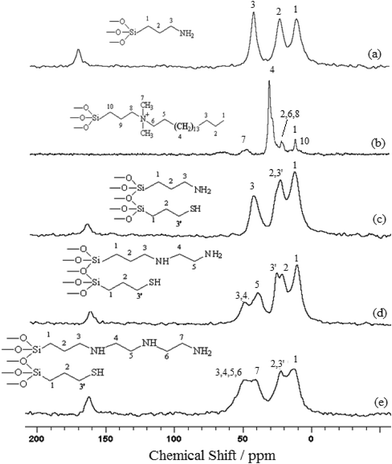 | ||
| Fig. 6 13C NMR spectra for the P-AT (a), P-OCT (b), P-ATM (c), P-EM (d) and P-DM (e) phyllosilicates. | ||
Three distinct signals at 14.4, 26.2 and 44.2 ppm for the P-AT spectrum were assigned to carbons 1, 2 and 3, respectively, as shown in Fig. 6a. For a long carbon chain, as observed for P-OCT phyllosilicate in Fig. 6b, there are overlapping peaks. In the case of P-ATM, shown in Fig. 6c, there is a similar spectrum to that obtained for P-AT, with three signals at 13.0, 23.7 and 43.3 ppm, related to carbons 1, 2 (3′) and 3, respectively. The carbon 3 signal, corresponding to the carbon linked to the mercapto functional group should appear around 28 ppm, but in this case it is observed that there were overlapping peaks with carbon 2. There are five signals at 13.5, 24.2, 28.4, 41.4 and 51.7 ppm for the P-EM spectrum in Fig. 6d that correspond to carbons 1, 2, 3′, 5 and 3(4), respectively. It is possible to observe the signal for the carbon linked to the mercapto group at 28.4 ppm and a new signal at 51.7 ppm, referring to 3 and 4 methylene groups, as shown in Fig. 6d. The spectrum of the P-DM, illustrated in Fig. 6e, shows four signals at 13.5, 23.7, 42.4 and 50.7 ppm assigned to carbons 1, 2(3′), 7 and (3, 4, 5, 6). In this case, the increase in carbon chain length caused a significant overlap of peaks.
For all compounds containing terminal amino groups the appearance of a peak near to 164 ppm, which was assigned to carbonyl carbon was observed. This peak may be related to the presence of sorbed carbon dioxide on the solid, since this gas can interact with amino groups to form carbamate species. This interaction is reversible, and the CO2 begins to be desorbed from 313 K.43
The thermogravimetric curves obtained for the phyllosilicate hybrids presented in Fig. 7 gave total mass losses of 42.2, 71.7, 42.0, 45.1 and 45.4% for P-AT, P-OCT, P-ATM, P-EM and P-DM, respectively. These total mass losses for all phyllosilicates are attributed to the combination of decomposition events of organic pendant chains from the attached silylating agents, in addition to the release of water molecules and formation of inorganic oxides, to end in the final residue.44 It was also observed that the size of the organic pendant chains directly affected the release, thus, the highest mass losses are in good agreement with elemental analysis results. An example is P-OCT that contains the largest chain and presented the highest mass loss, corresponding to 71.7%. In the case of bifunctional phyllosilicates, such as P-ATM, P-EM and P-DM the same behavior is also noted with an increase in total mass loss with the corresponding increase in chain length.
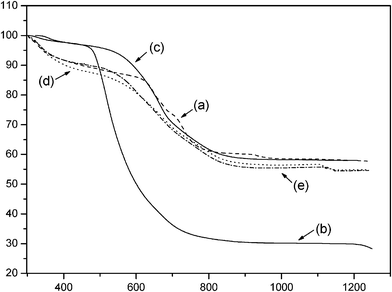 | ||
| Fig. 7 Thermogravimetric curve for P-AT (a), P-OCT (b), P-ATM (c), P-EM (d) and P-DM (e) phyllosilicates. | ||
The derivative thermogravimetric curves for the phyllosilicates presented, in all cases, three signals referring to mass losses, which are related in the first case, to the release of water molecules sorbed on the solid surfaces and also inside the lamellar cavity, in addition to molecules of solvent that were not completely removed during the drying process, while the second mass loss corresponds to the decomposition of the organic chains attached to the inorganic structures of the phyllosilicates. The third signal is due to condensation of the moieties containing oxygen or silanol groups formed during the decomposition, to form siloxane groups, therefore, the final collapse of the inorganic structure ended with oxide as residue. As an example, the derivative thermogravimetric curve for P-ATM phyllosilicate is shown in Fig. 8 and all other compounds presented the same general behavior.
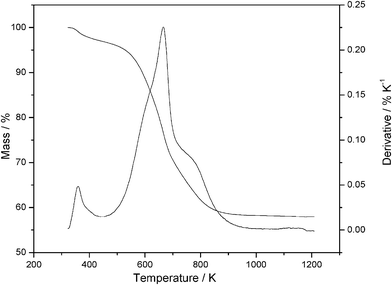 | ||
| Fig. 8 Thermogravimetric and derivative curves of the magnesium P-ATM phyllosilicate. | ||
The images for all magnesium phyllosilicates are very similar, corresponding to a planar morphology, suggesting the presence of several layers, as expected for a lamellar compound and a representative illustration is given for P-EM compound in Fig. 9.
 | ||
| Fig. 9 Scanning electron microscopy for P-EM phyllosilicate. | ||
Taking into account exploratory tests for dye sorption, it was observed that P-OCT and P-ATM presented the highest effectiveness in colorant removal. Therefore, these phyllosilicates were selected for the implementation of other sorption studies and for application in real samples of contaminated wastewater with dyes.
For all dye sorption isotherms a slight increase in pH was observed for the phyllosilicates, the optimum values of which are listed in Table 3. Based on the maximum capacity property RB can be more effectively removed by P-ATM, while P-OCT can be very useful to RY and RB, with a great advantage to using deionized water solution at pH very close to 6.
| Phyl | pH | Dye | Q m/mg g−1 | b/dm3 mg−1 | R 2 |
|---|---|---|---|---|---|
| P-ATM | 7.0 | RY | 735 | 0.005 | 0.995 |
| 6.1 | RB | 1043 | 0.003 | 0.996 | |
| 7.0 | RR | 549 | 0.005 | 0.985 | |
| P-OCT | 5.7 | RY | 1343 | 0.009 | 0.996 |
| 6.1 | RB | 1286 | 0.023 | 0.999 | |
| 7.0 | RR | 344 | 1.276 | 1.000 |
The isotherms showing time dependence were obtained for optimum pH values for each dye and the results are shown in Fig. 10. In examining the curves for RY dye in Fig. 10A, the equilibrium was reached after 700 min. For P-ATM and P-OCT with RB and RR in Fig. 10B and C, this time was much higher, in identical conditions for both dyes, occurring after 1440 min.
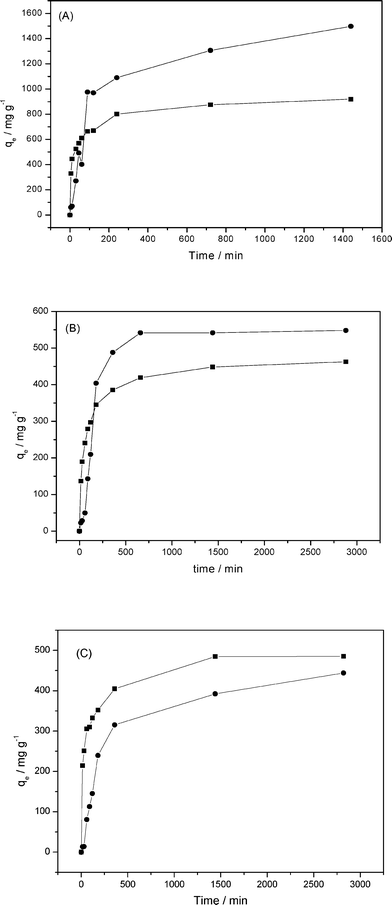 | ||
| Fig. 10 Influence of time on RY (A), RB (B) and RR (C) dye sorptions by P-OCT (●) and P-ATM (■) phyllosilicate. | ||
With the optimum pH conditions and defined contact time, sorption isotherms were obtained by varying the initial concentration and the results were fitted to the Langmuir model,45 which enables calculation of Qm values related to the maximum sorption capacity, as summarized in Table 3. From these results, P-OCT and P-ATM phyllosilicates gave very high maxima adsorption capacities. The first compound presented a higher sorption capacity for RY and RB dyes, even with the lowest functionalization degree. This degree of sorption could be associated with the fact that this phyllosilicate is already positively charged in the solid state due to the presence of the ammonium group, which facilitates electrostatic interactions with the anionic dye. In addition, this lamellar compound has a larger basal distance with long organic chains, containing eighteen carbon atoms, favoring the hydrophobic matrix/dye interactions through weak van der Waals type interactions, mainly due to having linear molecules.
It is interesting to observe that for phyllosilicate containing the OCT silylating agent, the highest values of sorption capacity were determined for RY, followed by RB and RR dyes. For bifunctionalized solids that have AT attached to the inorganic structure, there was an inversion of order between RB and RY dyes, with the highest value found for RB. These results demonstrate that the dye structures and the nature of the sorbents strongly influence the sorption processes. For example, RY dye is relatively planar or linear and presents a short carbon chain, which facilitates the diffusion of the dye inside the phyllosilicate structures and favors the hydrophobic interaction in the solid containing OCT agent. On the other hand, RB dye presents an an intramolecular interaction between SO3 and NH2 groups, both positioned in the benzene ring. Thus, the first group seems to cause a decrease of the effectiveness in the electrostatic interaction. Moreover, the carbon chain is more branched, hindering the diffusion and hydrophobic interactions. In this context, the RR dye has a more complex chemical structure and for this reason, the diffusion process, in this case, is more affected, reflected in the lower sorption obtained.44 These results suggest that the hydrophobic contribution is most important in solids containing OCT and electrostatic interactions in bifunctionalized phyllosilicates.
Comparing the sorption result capacities obtained for P-OCT with other sorbents previously reported, as shown in Table 4, it is clear that the present results show that the new phyllosilicates are more and more effective. It is interesting to observe that the experimental conditions and the dyes used are different in each case. Therefore, the results strongly indicate that the synthesized phyllosilicate is a promising removal of dyes, even those present in industrial effluents.
| Sorb | Dye | Q m/mg g−1 | Reference |
|---|---|---|---|
| P-OCT | RY GR | 1343 | This work |
| Activated sludge | RY 2 | 333 | 46 |
| Modified cotton | RY 23 | 302 | 47 |
| Chitosan | RY 145 | 188 | 11 |
| Sepiolite | RY 176 | 169 | 48 |
| Penicillium oxalicum | RY 145 | 137 | 49 |
| Fly ash | RY 84 | 37 | 9 |
| Mesoporous aminopropyl silica | RY, GR | 23 | 15 |
| P-OCT | RB, RN | 1286 | This work |
| Activated sludge | RB 2 | 250 | 46 |
| Chitosan | RB 222 | 199 | 50 |
| Penicillium oxalicum | RB 19 | 160 | 49 |
| Fly ash | RB 19 | 136 | 9 |
| P-OCT | RR RB | 344 | This work |
| Chitosan | RR 222 | 339 | 50 |
| Zeolite | RR, 239 | 111 | 48 |
| Fly ash | RR 198 | 47 | 9 |
| Penicillium oxalicum | RR 241 | 122 | 49 |
| Activated charcoal | BR, HE-3B | 120 | 51 |
| Mesoporous aminopropylsilica | RR, RB | 24 | 15 |
| Pine bark | RR 194 | 13 | 10 |
X-Ray diffraction patterns for P-AT after dye sorptions, shown for RY in Fig. 11Ab, have the peak for 2θ equal to 5.3° referring to the (001) plane, which corresponds to the basal distance, and show the disappearance of the peak at 2θ equal to 60° related to (060) plane, associated with the trioctahedral structure. This fact suggests that there was an exfoliation of the layered structure after dye interaction. For P-OCT, shown in Fig. 11B, there was a shift of the peak related to the (001) plane, from 2.6 to 2.1, 2.3 and 2.4°, respectively, for the RY, RB and RR dyes, showing an increase in the basal distance of 3.5 nm to 3.7, 3.7 and 4.2 nm, respectively. This result suggests that the dye molecule is interacting inside the interlayer space, remaining there and leading to an increase of the basal distance. Also the disappearance of the peak for 2θ equal to 60°, referring to the (060) plane, with broadening of the peak of 2θ equal to 20° referring to the (020) plane and the disappearance of peaks between 5 and 15°, indicates that there was a loss of a long-range order after dye sorption. However, the lamellar structure was preserved, as indicated by the intensity of (001) peak. For RR dye, that presented the smallest adsorption capacity, due to its nonlinear structure, some other peaks remain, indicating the lesser interaction between this dye and the sample.
 | ||
| Fig. 11 X-Ray diffraction patterns of P-AT (A-a) and P-OCT (B-a) before and after sorption of RA (b), RB (c) and RR (d) dyes. | ||
Based on the results obtained in the sorption isotherms, it was found that the phyllosilicate P-OCT showed better results for dye removal when compared to the others. After experimental optimization of conditions such as pH, contact time and mass, P-OCT was tested with three effluent samples collected from the textile industry. For this purpose, 0.50 g of P-OCT was suspended with 200 cm3 of effluent samples without adjustment of pH, for 360 min. The absorbance of the supernatant as a function of wavelength is shown in Fig. 12 and for all cases after treatment, the absorbance values decreased, being very close to null value. This result demonstrates that the phyllosilicate P-OCT is very promising as a sorbent for removal of these kinds of dyes. An illustration of this effective process is shown by pictures inserted in the absorbance curves, before and after the treatment samples. As observed, a significant change in color occurred after the treatment, resulting in almost colorless solutions at the end of the process. This visual analysis allows us to confirm once again the excellent performance of this phyllosilicate as a sorbent.
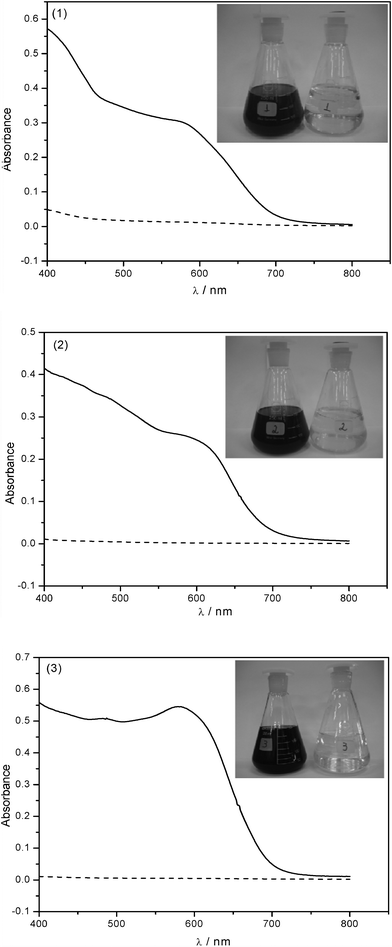 | ||
| Fig. 12 Sample of 200.0 cm3 of effluents 1, 2 and 3, using 500 mg of P-OCT, without adjustment of pH and contact time of 360 min under stirring. Effluent before (-) and after treatment (- - -) with the phyllosilicate. | ||
Some characteristics of the effluents were monitored before and after treatment in order to check for possible changes, indicating that the pH of the effluents remains the same after treatment. The differences of absorbance, turbidity and chemical oxygen demand (COD) values are listed in Table 5, with reduction of absorbance at all wavelengths, which confirms the efficiency of the treatment. The percentage reduction in the turbidity of the effluent after treatment gave high values of 98, 71 and 88%, for effluents 1, 2 and 3, respectively. Also the chemical oxygen demand (COD) values decrease to 35, 55 and 45% for the same sequence of effluents. Such reduction in COD shows that P-OCT phyllosilicate can remove the dye molecules, thereby decreasing the amount of organic matter contained in the analyzed effluents.
| Samp | 1 | 2 | 3 |
|---|---|---|---|
| ReA (417) | 93 | 98 | 98 |
| ReA (516) | 95 | 99 | 99 |
| ReA (590) | 96 | 99 | 99 |
| COD | 35 | 55 | 45 |
| Turb | 98 | 71 | 88 |
Conclusions
Magnesium phyllosilicates containing silylating agents in mono and bifunctional arrangements were successfully synthesized and characterized by several different techniques. These materials present 2:1 talc-like structure with an increase in the basal distance, in comparison with natural talc, data that are in good agreement with the size of the pendant organic groups attached. The inorganic framework presented high degrees of functionalization, proving the efficiency of the synthetic methodology, that shows both quantitatively and qualitatively the presence of T and absence of Q sites in 29Si NMR. The integrities of the organic chains for all materials were confirmed through 13C NMR determinations. The synthesized phyllosilicates demonstrated stability to over 500 K, which is good enough for their use as sorbents or even some different applications. The sorption experiments for reactive yellow GR, red RB and blue RN dyes, widely used in the textile industries, demonstrated that these phyllosilicates can be over a large pH range, with their optimum value near to 7 for yellow and red dyes and pH 6.1 for blue dye. The present results, especially for the phyllosilicates containing octadecyldimetyl(3-trimethoxysilylpropyl)-ammonium chloride, which has the highest sorption capacity, reveal potential use for dye removal in mild conditions, with a good, cheap agent, with great advantages in environmental applications.
Experimental
Reagents
Natural talc (T) was supplied by Magnesite SA, Brumado, BA, Brazil, with mass percentages of SiO2, Al2O3, Fe2O3, TiO2, CaO and MgO of 56.0, 1.5, 0.5, 0.05, 0.1 and 29.0%, respectively.The reagents sodium hydroxide, magnesium nitrate hexahydrate, hydrochloric acid, ethanol, 3-aminopropyltriethoxysilane (AT), 3-mercaptopropyltrimethoxysilane (M), 3-ethylenediaminopropyltrimethoxysilane (E), 3-diethylenetriaminopropyltrimethoxysilane (DT), octadecyldimethyl(3-trimethoxysilylpropyl)ammonium chloride (OCT) were used without purification.
The dyes Reactive Yellow GR (RY), Reactive Red RB (RR) and Reactive Blue RN (RB), supplied by Dystar Ltda, Suzano, SP, Brazil, were used without prior purification in deionized aqueous solutions. The structures of these dyes are shown in Fig. 2.
The effluent samples were collected from the textile dyeing industry from Americana, SP, Brazil. Three samples were collected on the same day, but at different times. These samples contain a mixture of reactive and dispersive dyes, hydrogen peroxide and caustic soda.
Synthesis and functionalization
To synthesize all magnesium phyllosilicates, the stoichiometric Si/Mg of 4/3 was maintained to provide the same ratio as found in natural talc.52 Initially, a solution containing 0.064 mol of the silylating agents AT or OCT in 150 cm3 of ethanol was dropped into 0.048 mol of Mg(NO3)2·6H2O in 150 cm3 of ethanol, in an oil bath at 313 K. To the hot mechanically stirred mixture was added 0.096 mol of sodium hydroxide from a 0.50 mol dm−3 solution. The suspension formed was stirred for 24 h and aged for 7 days. The solid was centrifuged, washed with ethanol and deionized water to pH 7, to yield the phyllosilicates P-AT or P-OCT. The syntheses of the binary combination of silylating agents AT-M, E-M and D-M yielded P-ATM, P-EM and P-DM phyllosilicates. In these preparations the required amount of silylating agents (0.064 mol) was equivalently divided between the two components of the specific phyllosilicate. The structures of all pendant organic groups attached to the synthesized phyllosilicates are illustrated in Fig. 1.Characterization
The functionalization degree of the synthesized compounds was determined by the quantities of nitrogen, hydrogen and carbon in a Perkin Elmer model PE 2400 elemental analyzer. The infrared spectra of the samples in KBr pellets were obtained by accumulating 32 scans on a Bomem Spectrophotometer, MB-series, in the 4000 to 400 cm−1 range with 4 cm−1 of resolution. X-Ray diffraction (XRD) patterns were obtained on a Shimadzu XD7000 diffractometer (40 kV, 30 mA), in the 2θ from 1.4 to 70° range with nickel-filtered Cu-Kα radiation, with a wavelength of 0.154 nm. Solid state NMR spectra for carbon and silicon nuclei for all samples were obtained on a Bruker AC 300/P spectrometer at room temperature. For each run, approximately one gram of each solid sample was compacted into a 7 mm zirconium oxide rotor. The measurements were obtained at frequencies of 75.47 and 59.61 MHz, with a magic angle spinning of 4 kHz. The contact times of 1 and 3 ms and time of repetition of 1 and 3 s were used for carbon and silicon, respectively. The thermogravimetric curves in nitrogen atmosphere were obtained on a DuPont instrument, model 9900, using heating rates of 0.17 K s−1, varying from room temperature to 1273 K. Scanning electron microscopy (SEM) data were obtained from detection of the secondary electron images on a JEOL JSM 6360LV scanning electron microscope, operating at 20 kV. The samples were supported in the sample holder containing a copper ribbon. After preparation, the samples were subjected to gold plating and were then analyzed. The pH measurements were performed on a pH/Ion Model 450 M pH meter, using a combined glass electrode. For determination of dye concentrations a Shimadzu spectrophotometer model MultiSpec-1501 TCC-240A was used. The wavelengths of maximum absorption of each dye were 417, 516 and 590 nm for Reactive Yellow GR, Red RB and Blue RN, respectively.Sorption
For all synthesized phyllosilicates used in the sorption investigations, the values obtained were compared with unfunctionalized natural talc. Early tests were performed to follow the dye sorption behavior, which consisted in stirring flasks with masses near to 20 mg of each phyllosilicate, that were added to solutions of each dye. The flasks were maintained under orbital stirring at 298 ± 1 K for 24 h and after filtration, the solutions were diluted, when necessary, to determine the absorbance.Influence of pH
Stock solutions of 20.0 g dm−3 for each dye were prepared, which were diluted with buffer solutions of the following pH 4, 7 and 9, to obtain final solutions of 4.0 g dm−3 in concentrations. For these determinations a series of flasks containing about 20 mg of phyllosilicates T-, P-ATM and P-OCT were placed in contact with 6.0 cm3 of these solutions with the series of pH. The same procedure was performed as before and the dye concentrations were determined in the filtered fractions for each dye.Influence of time
The influence of time on the sorptions was determined through a series of flasks with masses near to 20 mg of the phyllosilicates P-ATM and P-OCT and placed in contact with 6.0 cm3 solution of 4.0 g dm−3 of each dye at optimum pH. The flasks were maintained under orbital stirring from 5 to 1440 min for yellow dye, 5 to 2880 min for blue and red dyes. After filtration, the final concentrations were also determined as before.Sorption isotherm
A series of flasks containing around 20 mg of P-ATM and P-OCT phyllosilicates were suspended in contact with 6.0 cm3 of solution of different concentrations ranging from 100 to 20000 mg dm−3 of each dye. These flasks were stirred before and after filtration, and the final concentrations were determined. In all cases the sorbed amounts were calculated according to eqn (1):| qe = (Ci − Ce)V/w | (1) |
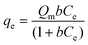
| (2) |
Sorption in real effluent samples
Initially, samples of around 20 mg of the synthesized P-AT, P-OCT, P-ATM, P-EM and P-DM phyllosilicates were suspended with 6.0 cm3 of each effluent. The suspensions were maintained under orbital stirring for 24 h at 298 ± 1 K, when the solid was filtered and the supernatant had the absorbance recorded. The same procedure was performed with pH adjusted to 7 for each effluent, by adding 0.10 mol dm−3 hydrochloric acid solution. To determine the influence of the contact time, the same procedure was again performed with P-OCT phyllosilicate, ranging from 0.5 to 24 h. In addition, the influence of the mass was also checked with a variation from 0.005 to 0.020 g.Following optimization of the experimental conditions such as pH, contact time and influence of mass, phyllosilicates were used in treatment of real wastewater samples. For this purpose, phyllosilicate masses around 500 mg were suspended with 200.0 cm3 of each effluent. After stirring for 6 h, the solid was filtered and the absorbance of the supernatant was recorded in the 400 to 800 nm range. From this range the absorbances at 417, 516 and 590 nm were considered, taking into account that these are the corresponding wavelengths for the dyes used in this investigation, the calculated efficiency for each of them was obtained from eqn (3):
| E(%) = (Ao − A/Ao) × 100 | (3) |
Acknowledgements
The authors are indebted to FAPESP and CNPq for fellowship and financial support.References
- N. M. Mahmoodi, B. Hayati, M. Arami and H. Bahrami, Desalination, 2011, 275, 93–101 CrossRef CAS.
- V. K. Gupta and Suhas, J. Environ. Manage., 2009, 90, 2313–2342 CrossRef CAS.
- N. Durán, S. G. Morais and R. S. Freire, Chemosphere, 2000, 40, 369–373 CrossRef.
- Y. Ozdemir, M. Dogan and M. Alkan, Microporous Mesoporous Mater., 2006, 96, 419–427 CrossRef.
- Z. Aksu, Process Biochem., 2005, 40, 997–1026 CrossRef CAS.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS.
- D. C. W. Tsang, J. Hu, M. Y. Liu, W. Zhang, K. C. K. Lai and I. M. C. Lo, Water, Air, Soil Pollut., 2007, 184, 141–155 CrossRef CAS.
- A. R. Dincer, Y. Gunes, N. Karakaya and E. Gunes, Bioresour. Technol., 2007, 98, 834–839 CrossRef CAS.
- N. Dizge, C. Aydiner, E. Demirbas, M. Kobya and S. Kara, J. Hazard. Mater., 2008, 150, 737–746 CrossRef CAS.
- E. C. Lima, B. Royer, J. C. P. Vaghetti, N. M. Simona, B. M. Cunha, F. A. Pavan, E. V. Benvenuttia, R. Cataluña-Veses and C. Airoldi, J. Hazard. Mater., 2008, 155, 536–550 CrossRef CAS.
- M. S. Chiou and H. Y. Li, Dyes Pigm., 2004, 60, 69–84 CrossRef CAS.
- A. P. Vieira, S. A. A. Santana, C. W. B. Bezerra, H. A. S. Silva, J. A. P. Chaves, J. C. P. Melo, E. C. Silva Filho and C. Airoldi, J. Hazard. Mater., 2009, 166, 1272–1278 CrossRef CAS.
- A. P. Vieira, S. A. A. Santana, C. W. B. Bezerra, H. A. S. Silva, J. A. P. Chaves, J. C. P. Melo, E. C. Silva Filho and C. Airoldi, Chem. Eng. J., 2011, 173, 334–340 CrossRef CAS.
- A. Safa Ozcan, B. Erdem and A. Ozcan, Colloids Surf., A, 2005, 266, 73–81 CrossRef.
- A. R. Cestari, E. F. S. Vieira, G. S. Vieira and L. E. Almeida, J. Colloid Interface Sci., 2007, 309, 402–411 CrossRef CAS.
- R. Guegan, M. Gautier, J.-M. Beny and F. Muller, Clays Clay Miner., 2009, 57, 502–509 CrossRef CAS.
- B. Royer, N. F. Cardoso, E. C. Lima, T. R. Macedo and C. Airoldi, J. Hazard. Mater., 2010, 181, 366–374 CrossRef CAS.
- B. Royer, N. F. Cardoso, E. C. Lima, T. R. Macedo and C. Airoldi, Sep. Sci. Technol., 2010, 45, 129–141 CrossRef CAS.
- M. Jaber, J. Miehé-Brendlé, L. Michelin and L. Delmotte, Chem. Mater., 2005, 17, 5275–5281 CrossRef CAS.
- T. R. Macedo, G. C. Petrucelli and C. Airoldi, Thermochim. Acta, 2010, 502, 30–34 CrossRef CAS.
- T. R. Macedo and C. Airoldi, New J. Chem., 2009, 33, 2081–2089 RSC.
- S. Badshah and C. Airoldi, Chem. Eng. J., 2011, 166, 420–427 CrossRef CAS.
- M. A. Melo Jr and C. Airoldi, Dalton Trans., 2010, 39, 10217–10227 RSC.
- L. Zampori, P. Gallo Stampino and G. Dotelli, Appl. Clay Sci., 2009, 42, 605–610 CrossRef CAS.
- Y. Gonen and G. Rytwo, J. Colloid Interface Sci., 2006, 299, 95–101 CrossRef CAS.
- J.-C. Gallégo, M. Jaber, J. Miehé-Brendlé and C. Marichal, New J. Chem., 2008, 32, 407–412 RSC.
- R. Sasai, H. Itoh, I. Shindachi, T. Shichi and K. Takagi, Chem. Mater., 2001, 13, 2012–2016 CrossRef CAS.
- T.-Y. Chao, H.-L. Chang, W.-C. Su, J.-Y. Wu and R.-J. Jeng, Dyes Pigm., 2008, 77, 515–524 CrossRef CAS.
- M. Borisover, E. R. Graber, F. Bercovich and Z. Gerstl, Chemosphere, 2001, 44, 1033–1040 CrossRef CAS.
- M. G. Fonseca and C. Airoldi, J. Chem. Soc., Dalton Trans., 1999, 21, 3687–3692 RSC.
- D. L. Pavia, G. M. Lampman, G. S. Kriz, Introduction to Spectroscopy, 2nd edn, Saunders, New York, 1996 Search PubMed.
- M. G. Fonseca, J. S. Barone and C. Airoldi, Clays Clay Miner., 2000, 48, 638–647 Search PubMed.
- S. L. Burkett, A. Press and S. Mann, Chem. Mater., 1997, 9, 1071–1073 CrossRef CAS.
- K. Fujii, S. Hayashi and H. Kodama, Chem. Mater., 2003, 15, 1189–97 CrossRef CAS.
- A. Worayingyoung, P. Kangvansura, S. Ausadasuk and P. Praserthdam, Colloids Surf., A, 2008, 315, 217–225 CrossRef.
- T. Vralstad, H. K. Magnusson and J. Sjöblom, Microporous Mesoporous Mater., 2007, 106, 155–161 CrossRef CAS.
- B. Velde, Introduction to Clay Minerals, Chapman & Hall, London, 1992 Search PubMed.
- H. Hussain, H. U. Rehman and Z. Ahmad, J. Sol-Gel Sci. Technol., 2005, 36, 239–248 CrossRef CAS.
- K. Fujii, S. Hayashi and H. Kodama, Chem. Mater., 2003, 15, 1189–1197 CrossRef CAS.
- M. Jaber, J. M. Brendlé, L. Delmotte and R. L. Dred, Solid State Sci., 2005, 7, 610–615 CrossRef CAS.
- N. T. Whilton, S. L. Burkett and S. Mann, J. Mater. Chem., 1998, 8, 1927–1932 RSC.
- J. A. A. Sales, G. C. Petrucelli, F. J. V. E. Oliveira and C. Airoldi, J. Colloid Interface Sci., 2006, 297, 95–103 CrossRef CAS.
- M. Rocchia, E. Garrone, F. Geobaldo, L. Boarino and M. J. Sailor, Phys. Status Solidi A, 2003, 197, 365–369 CrossRef CAS.
- A. R. Cestari, E. F. S. Vieira, A. G. P. Santos, J. A. Mota and V. P. Almeida, J. Colloid Interface Sci., 2004, 280, 380–386 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1368 CrossRef CAS.
- Z. Aksu, Biochem. Eng. J., 2001, 7, 79–84 CrossRef CAS.
- I. Bouzaida and M. B. Rammah, Mater. Sci. Eng., C, 2002, 21, 151–155 CrossRef.
- O. Ozdemir, B. Armagan, M. Turan and M. S. Celik, Dyes Pigm., 2004, 62, 49–60 CrossRef CAS.
- S. J. Zhang, M. Yang, Q. X. Yang, Y. Zhang, B. P. Xin and F. Pan, Biotechnol. Lett., 2003, 25, 1479–1482 CrossRef CAS.
- F. Wu, R. Tseng and R. Juang, J. Hazard. Mater., 2001, 81, 167–174 CrossRef CAS.
- D. Suteu and D. Bilba, Acta Chim. Slov., 2005, 52, 73–79 CAS.
- A. S. O. Moscofian and C. Airoldi, J. Hazard. Mater., 2008, 160, 63–6 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |