Electronic and molecular structures and bulk second–order nonlinear optical properties of ferrocenyl ynones†
Damian
Plażuk
a,
Janusz
Zakrzewski
*a,
Keitaro
Nakatani
b,
Anna
Makal
c,
Krzysztof
Woźniak
c and
Sławomir
Domagała
d
aDepartment of Organic Chemistry, University of Łódź, Tamka, 12, 91-403, Łódź, Poland
bPPSM, ENS Cachan, CNRS, UniverSud, 61 av President Wilson, 94230, Cachan, France
cDepartment of Chemistry, Warsaw University, Pasteura, 1, 02-093, Warszawa, Poland
dDepartment of Inorganic and Analytical Chemistry, University of Łódź, Tamka, 12, 91-403, Łódź, Poland
First published on 2nd March 2012
Abstract
Ferrocenyl ynones FcCOC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR (R = H, TMS, Ph) exhibit moderate second harmonic generation efficiencies in the solid state, whereas the methyl analog proved inactive. Their molecular structures and crystal packings were determined by single crystal X-ray diffraction, confirming that the inactive compound crystallizes in a centrosymmetric space group. The electronic structures of these compounds were studied experimentally (cyclic voltammetry, electronic absorption spectra) and theoretically (DFT and TD-DFT calculations). It appeared that the COCCR groups are stronger electron acceptors than the propionyl (COCH2CH3) group. Furthermore, these groups better stabilize metal-centred HOMO-2 to HOMO orbitals and lateral chain-centred LUMO orbitals and decrease HOMO-LUMO gaps. The TDDFT calculation of electronic transitions revealed that lower energy (LE) and higher energy (HE) bands observed in the electronic absorption spectra of compounds under study have more pronounced metal-to-ligand charge transfer character for ferrocenyl ynones than for propionylferrocene. The calculated static quadratic hyperpolarizabililities of ferrocenyl ynones are in the range ∼3 − 6 × 10−30 esu.
CR (R = H, TMS, Ph) exhibit moderate second harmonic generation efficiencies in the solid state, whereas the methyl analog proved inactive. Their molecular structures and crystal packings were determined by single crystal X-ray diffraction, confirming that the inactive compound crystallizes in a centrosymmetric space group. The electronic structures of these compounds were studied experimentally (cyclic voltammetry, electronic absorption spectra) and theoretically (DFT and TD-DFT calculations). It appeared that the COCCR groups are stronger electron acceptors than the propionyl (COCH2CH3) group. Furthermore, these groups better stabilize metal-centred HOMO-2 to HOMO orbitals and lateral chain-centred LUMO orbitals and decrease HOMO-LUMO gaps. The TDDFT calculation of electronic transitions revealed that lower energy (LE) and higher energy (HE) bands observed in the electronic absorption spectra of compounds under study have more pronounced metal-to-ligand charge transfer character for ferrocenyl ynones than for propionylferrocene. The calculated static quadratic hyperpolarizabililities of ferrocenyl ynones are in the range ∼3 − 6 × 10−30 esu.
1. Introduction
Electronic and optical properties of conjugated ferrocene and, to a lesser extent, ruthenocene derivatives, have been extensively studied experimentally and theoretically1–5 due to their potential applications in non-linear optics and molecular electronics.6–8 Special attention has been paid to strongly polarized ferrocenyl-π-bridge-strong acceptor systems, exhibiting very large dipole moments and quadratic hyperpolarizabilities.9–12 However, as a rule, these compounds crystallize in centrosymmetric space groups with antiparallel alignment of NLO-dipoles, resulting in vanishing of bulk second-order NLO properties such as second harmonic generation (SHG). From this point of view, molecules bearing weaker acceptor groups, having smaller dipole moments, which may exhibit higher tendency to crystallization in non-centrosymmetric space group are of obvious interest. An example of such a compound is (Z)-1-ferrocenyl-2-(p-nitrophenyl)ethylene 1.13 This compound displays only a modest quadratic hyperpolarizability but has a relatively high SHG efficiency (62 × urea) due to formation of non-centrosymmetric crystals (interestingly, the (E)-isomer of 1, exhibiting higher quadratic hyperpolarizabity, forms SHG-inactive centrosymmetric crystals).14 Other examples of SHG-active crystals of compounds featuring this chromophore have also been reported.15Recently we reported synthesis of ferrocenyl ynones 2a–d.16 Ferrocenyl ketones are weak donor-acceptor systems exhibiting only very small quadratic hyperpolarizabilities (∼0.3 × 10−30 esu).6 Nevertheless, we thought that it would be of interest to check whether crystals of 2a–d exhibit SHG activity. Pleasingly, we found that three of four compounds under study showed SHG efficiencies 2–5 × urea. This prompted us to study their electronic and molecular structures and crystal packing in order to understand their mutual relationship. Furthermore, we performed DFT calculations on 2a–d and, for comparison, on their saturated analog, propionylferrocene 3. The data obtained, reported in this paper, casts new light on the electronic structure of conjugated ferrocenyl systems.
2. Results and discussion
2. 1. Electronic absorption spectra
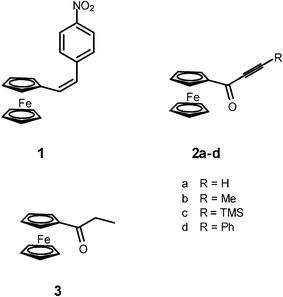
Electronic absorption spectra of 2a–d and, for comparison, the spectrum of a saturated ferrocenyl ketone, propionylferrocene 3, were measured in chloroform solutions at room temperature. The spectra are shown in Fig. 1 and the spectroscopic data are collected in Table 1. | ||
| Fig. 1 Electronic absorption spectra of 2a–d and 3 in chloroform. | ||
| Compound | HE band | LE band | ||
|---|---|---|---|---|
| λ max/nm | ε max/M−1cm−1 | λ max/nm | ε max/M−1cm−1 | |
| 2a | 375 | 1600 | 492 | 1200 |
| 2b | 368 | 1600 | 482 | 1000 |
| 2c | 377 | 1800 | 493 | 1400 |
| 2d | 366 | 1800 | 497 | 1200 |
| 3 | 335 | 1200 | 454 | 400 |
Similarly as other ferrocenyl derivatives, compounds 2a–d and 3 display in the near UV-Vis region of their electronic absorption spectra two bands, denoted as high energy (HE) and low energy (LE) bands.2,17 These bands are usually assigned to ligand field d-d transitions with some admixture of metal-to-ligand charge transfer (MLCT) transitions. For ferrocenyl ketones this means that the contribution of the charge-separated resonance structure 4 is more important in the excited state than in the ground state.17
It can be seen from Table 1 and Fig. 1 that the HE and LE bands of 2a–d are shifted bathochromically and are more intense than the corresponding bands of 3. This suggests a stronger admixture of the MLCT transition to both transitions for acetylenic ketones.
2.2. Electrochemistry
Cyclic voltammetry technique was used for study of the redox properties of 2a–d. The measurements were carried out in dichloromethane solutions in the range of potentials expected for the redox changes involving the ferrocene/ferrocenium couple. The data are shown in Table 2, which also contains, for comparison, the data for 3.| Compound | E 1/2 (mV) vs. Fc+/Fc | ΔE (mV) |
|---|---|---|
| 2a | 335 | 102 |
| 2b | 292 | 106 |
| 2c | 328 | 124 |
| 2d | 316 | 111 |
| 3 | 248 | 133 |
All compounds under study exhibit quasi-reversible oxidation/reduction of the ferrocenyl moiety. Their redox potential are shifted anodically in comparison to that of ferrocene, consistently with the electron-withdrawing nature of acyl substituents. Alkynoyl substituents present in 2a–d exert stronger electron-withdrawing effect than the saturated propionyl substituent in 3. This is in contrast to the reported behavior of an alkenoyl ferrocene, which is more readily oxidizable than acetylferrocene, a phenomenon which was tentatively explained assuming that the electron attracting power of the carbonyl group decreases as a result of conjugation with the ethylenic bond.18 The reverse effect observed for 2a–d suggests that the electron-withdrawing power of the carbonyl group bound to an alkynyl group is higher than that of the group bound to an alkyl moiety. In our opinion, this may be explained by a higher polarization expected for a sp2-sp C–C bond in a O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CC entity due to a higher electronegativity of the sp carbons.
C–CC entity due to a higher electronegativity of the sp carbons.
The values of redox potentials reveal better stabilization of HOMO in 2a–d in comparison to 3. This has been confirmed by DFT calculations (vide infra).
2.3. Second harmonic generation
The bulk SHG efficiencies of complexes 2a–d have been measured by the Kurtz–Perry test at λ = 1907 nm. The results are collected in Table 3. Compounds 2a,c,d, show SHG efficiencies, whereas compound 2b was inactive, suggesting centrosymmetric space group (vide infra). It is worth noting that all compounds studied are very stable and their SHG efficiencies did not change after a 5 month stocking between two glass plates at room temperature.Although numerous ferrocenyl derivatives exhibit large quadratic hyperpolarizabilities,1–6 compounds in this class showing enhanced SHG efficiencies (> that of urea) in the crystalline state remain still rather rare.1,8,9 Compounds 2a,c,d show moderate SHG efficiencies but their advantage is a very simple synthesis (one step from commercially available and cheap ferrocene) and good stability.
2. 4. X-ray diffraction study
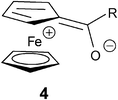
Only compound 2b crystallizes in a common P21/n centrosymmetric space group in a monoclinic system. It is worth mentioning, however, that the β angle in this case is close to 90 degrees. Compound 2a crystallizes in the chiral P212121 space group, whereas compounds 2c and 2d both crystallize in the polar Pna2(1) space group. In each case the crystals contain a single molecule compound, located in a general position in the crystallographic asymmetric unit without a solvent. The ORTEP representations of their structures are presented in the Fig. 2. | ||
| Fig. 2 ORTEP representations of 2a–d. The thermal ellipsoids were drawn at 50% probability level. | ||
In all structures an eclipsed conformation of the ferrocenyl moiety is observed, with exact ϕ angle values presented in Table 4. The ϕ angle is defined here as a C(1)–cent1–cent2–C(6) dihedral angle (cent1 and cent2 are the centroids of C(1)–C(5) and C(6)–C(10) rings, respectively). 2c adopts the ϕ angle closest to 0, while the phenyl derivative displays the deviation from eclipsed conformation by more than 5 degrees.
| 2a | 2b | 2c | 2d | |||||
|---|---|---|---|---|---|---|---|---|
| average Fe–C | 2.045 | (11) | 2.047 | (10) | 2.046 | (14) | 2.046 | (9) |
| ring C(1)–C(5) | 2.044 | (16) | 2.045 | (14) | 2.045 | (2) | 2.044 | (13) |
| ring C(6)–C(10) | 2.047 | (4) | 2.048 | (3) | 2.047 | (8) | 2.048 | (3) |
| Fe-cent1 | 1.644 | (2) | 1.645 | (2) | 1.645 | (2) | 1.644 | (2) |
| Fe-cent2 | 1.655 | (2) | 1.653 | (2) | 1.652 | (2) | 1.652 | (2) |
| average C–C bond | 1.427 | (11) | 1.428 | (7) | 1.429 | (12) | 1.429 | (10) |
| average C–C | 1.416 | (5) | 1.423 | (3) | 1.420 | (3) | 1.423 | (2) |
| average C–C–C angle | 108.0 | (3) | 108.0 | (3) | 108.0 | (6) | 108.0 | (5) |
| cent1-Fe(1)-cent(2) | 178.19 | (13) | 178.76 | (18) | 178.02 | (18) | 178.82 | (15) |
| ϕ | −2.06 | (14) | 3.5 | (2) | 0.66 | (15) | 4.93 | (16) |
| O(1)–C(11) | 1.2277 | (18) | 1.230 | (2) | 1.226 | (3) | 1.226 | (18) |
| C(1)–C(11) | 1.453 | (2) | 1.466 | (2) | 1.460 | (3) | 1.462 | (2) |
| C(11)–C(12) | 1.461 | (2) | 1.452 | (2) | 1.461 | (3) | 1.4608 | (19) |
| C(12)–C(13) | 1.185 | (2) | 1.196 | (2) | 1.205 | (3) | 1.196 | (2) |
| C(2)–C(1)–C(11) | 125.73 | (13) | 124.50 | (15) | 124.01 | (18) | 124.08 | (12) |
| C(5)–C(1)–C(11) | 125.80 | (13) | 127.31 | (15) | 127.46 | (18) | 127.64 | (12) |
| O(1)–C(11)–C(1) | 123.59 | (14) | 121.63 | (16) | 122.39 | (18) | 122.55 | (13) |
| O(1)–C(11)–C(12) | 120.57 | (14) | 121.34 | (15) | 119.97 | (18) | 120.42 | (13) |
| C(13)–C(12)–C(11) | 178.41 | (18) | 179.33 | (19) | 176.4 | (2) | 177.43 | (15) |
| C(12)–C(13)–X | 179.97 | (18) | 178.60 | (18) | 176.0 | (2) | 177.81 | (15) |
| C(11) out of C(1)–C(5) ring plane | −171.56 | (13) | −177.49 | (15) | −174.95 | (18) | −176.50 | (12) |
| C(2)–C(1)–C(11)–O(1) | −13.0 | (2) | −6.9 | (3) | −15.5 | (3) | −8.3 | (2) |
The Cp ligands are nearly parallel, as the angle between their planes does not exceed 2 degrees for any of the presented structures.
The C–C distances in the Cp rings tend to be longer for a substituted carbon (C(1) carbon in all the compounds) by about 0.01–0.02 Å, whereas the C(1)–Fe distance is the shortest in all cases, by about 0.02 Å. Either C(2) or C(5) also tend to be closer than the average distance to the iron atom. The substituted Cp ring is also closer to the iron atom by about 0.01 Å, as measured by the Fe distances to the centroids of the C(1)–C(5) and C(6)–C(10) rings.
In all compounds the alkynoyl groups are bent towards the middle of the sandwich complex, which is indicated by the negative values of C(11)–C(1)–C(2)–C(3) angles (Table 1). The carbonyl oxygen is twisted away from the sandwich center. The alkynyl fragment is not ideally linear, as indicated by appropriate angles in table. Therefore, all the substituents are to some extent inclined toward the middle of the ferrocenyl sandwich. The deviation from linearity, measured by a root mean square difference from 180 degrees of valence angles adjacent to the triple bond, roughly depends on the size of substituents. It is the smallest for 2b, slightly larger for 2a, and significantly larger for 2d and the highest for 2c. The phenyl ring in 2d is twisted by 45.81(8) degrees from the plane of the carbonyl group, and the hydrogen H19 is directed toward the centre of the ferrocenyl moiety and is actually located closer to the unsubstituted Cp ring (the distances to the closest H atoms from substituted–H(2)– and unsubstituted–H(7)–Cp rings are 4.013(3) Å and 3.868(4) Å, respectively).
Compound 2a shows an interesting network of C–H⋯O contacts. Only two such interactions are found, both of them nearly perpendicular to the plane of carbonyl moiety, in which the charge concentrations of lone electron pairs of the oxygen atom should be located. The first is quite short C–C–H⋯O contact of H(13) with O(1) atom from a molecule related by 2(1)[100] screw axis. The C(13)–H(13)⋯O(1) angle is about 157 degrees, which means that the whole alkynyl moiety is at nearly straight angle with respect to the carbonyl plane. A network of such contacts results in a column of 2a molecules being built along {100} direction. The second short contact, from above the carbonyl plane is an C(5)–H(5)⋯O(1) interaction between molecules related by 2(1)[010] screw axis. The remaining short contacts are all of the C–H⋯π type, engaging hydrogen and carbon atoms from the neighboring Cp rings. Fig. 3 illustrates the packing motifs and a packing diagram of the structure of 2a. Detailed description of C–H⋯O contacts geometry is summarized in Table 5. It appears that a chiral P212121 space group provides the effective, dense packing only in the case of molecules which are not significantly elongated.
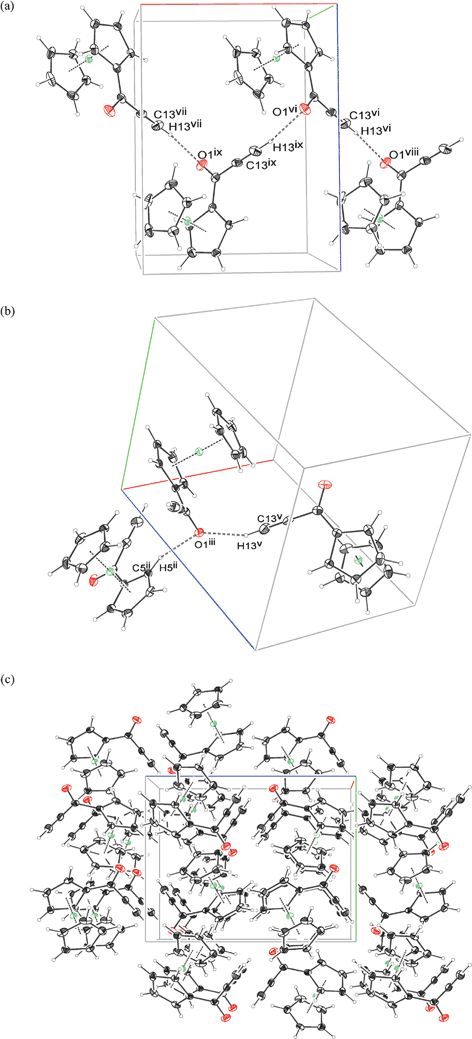 | ||
| Fig. 3 The most important intermolecular interactions (a, b) and crystal packing diagram in 100 direction (c) for the structure of 2a. Symmetry codes: subfigure (a) (ii) −1 + x, −1 + y, z ; (iii) 1 − x, −1/2 + y, 1/2 − z ; (v) 3/2 − x, 1 − y, 1/2 + z ; subfigure (b) (vi) 1 − x, −1/2 + y, 1/2 − z ; (vii) 2 − x, −1/2 + y, 1/2 − z ; (viii) 1/2 − x, 1 − y, 1/2 + z ; (ix) 3/2 − x, 1 − y, 1/2 + z. | ||
| D–H⋯A | d D–H | d H⋯A | d D⋯A | < D–H⋯A | symmop A | d H⋯A – ΣVdW | |
|---|---|---|---|---|---|---|---|
| 2a | C13–H13⋯O1 | 0.95 | 2.20 | 3.098(2) | 156.8 | −0.5 + x,1.5 − y,−z | −0.52 |
| C5–H5⋯O1 | 1.00 | 2.51 | 3.2030(18) | 129.8 | 2 − x,0.5 + y,0.5 − z | −0.21 | |
| 2b | C14–H14C⋯O1 | 0.98 | 2.41 | 3.313(2) | 153.6 | 1 − x,−y,−z | −0.31 |
| C14–H14A⋯O1 | 0.98 | 2.64 | 3.388(2) | 133.8 | 1 + x,y,z | −0.08 | |
| C5–H5⋯O1 | 1.00 | 2.64 | 3.527(2) | 147.2 | 1 + x,y,z | −0.08 | |
| C3–H3⋯O1 | 1.00 | 2.57 | 3.353(2) | 135.5 | 0.5 + x,0.5 − y,0.5 + z | −0.15 | |
| 2c | C15–H15A⋯O1 | 0.98 | 2.59 | 3.405(2) | 142.4 | x,y,1 + z | −0.13 |
| C2–H2⋯O1 | 1.00 | 2.60 | 3.467(2) | 147.8 | x,y,−1 + z | −0.12 | |
| C4–H4⋯O1 | 1.00 | 2.59 | 3.338(2) | 133.2 | 1.5 − x,−0.5 + y,−0.5 + z | −0.13 | |
| 2d | C19–H19⋯O1 | 1.00 | 2.53 | 3.3122(18) | 139.6 | x,1 + y,z | −0.19 |
| C7–H7⋯O1 | 1.00 | 2.58 | 3.380(2) | 141.7 | x,−1 + y,z | −0.14 | |
| C2–H2⋯O1 | 1.00 | 2.62 | 3.414(2) | 140.9 | x,−1 + y,z | −0.10 |
In crystals of 2b two of the three methyl hydrogens are engaged in close contacts with the carbonyl oxygen. The interactions result in formation of rings around symmetry centers, constituted in the case of first interaction by anti-parallelly located propynyl moieties (the short C–C distances are 4.144(2) Å in the case of alkynyl C12⋯C13 carbon atoms and 4.162(2) Å in the case of C11⋯C14 atoms). The second interaction is constituted by two methyl groups and two carbonyl oxygens from four surrounding molecules. In the latter case, the hydrogens from two methyl groups are very close to each other. This results in stacks of methyl to methyl oriented 2b molecules being built along the {100} direction. The remaining methyl hydrogen and the hydrogens from Cp rings are directed towards the nearest Cp planes, resulting in packing illustrated in the Fig. 4. Both oxygen and the carbon atoms involved in triple bond are exposed to additional interactions with H(3) and H(4) hydrogens from the closest perpendicular Cp ring respectively. The anti-parallel close orientation of the triple bond containing fragments may explain why the moiety in the case of DPynMe is the least distorted from linearity.
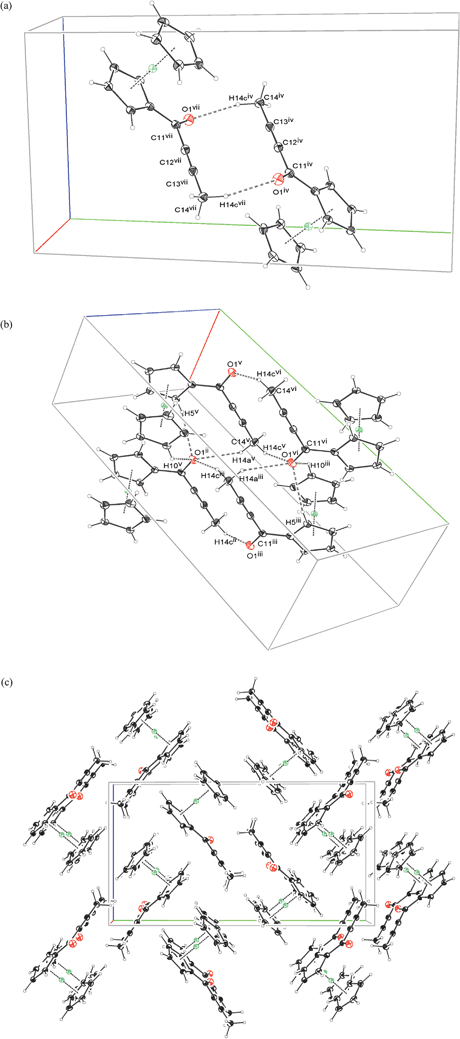 | ||
| Fig. 4 The most important interactions (a, b) and crystal packing diagram in 100 direction (c) for the structure of 2b. Symmetry codes (a): (iv) 3/2 − x, 1/2 + y, 1/2 − z ; (vii) 1/2 + x, 1/2 − y, 1/2 + z ; Symmetry codes (b): (ii) 1/2 + x, 1/2 − y, 1/2 + z ; (iii) 3/2 − x, 1/2 + y, 1/2 − z ; (iv) 1 + x, y, 1 + z ; (v) −1/2 + x, 1/2 − y, 1/2 + z. | ||
It appears that the methyl group with its three symmetrically located hydrogen atoms enables the dimers to form, and, consequently, centrosymmetric packing.
Compounds 2c and 2d present a great similarity in crystal packing. The reason for their adopting the same space group and similar cell volumes is the main packing motif represented by both molecules.
Their bulk molecular sizes are similar, and both form a hydrogen-reach cavity between the ferrocenyl moiety and a bulky substituent, to which an exposed oxygen from a carbonyl group has easy access. As an effect, stacks of parallel oriented and closely fitted molecules related by crystallographic translation are formed, as presented in the Fig. 5a and 5b, along the shortest crystallographic axis, which is b and c in the case of 2c and 2d, respectively.
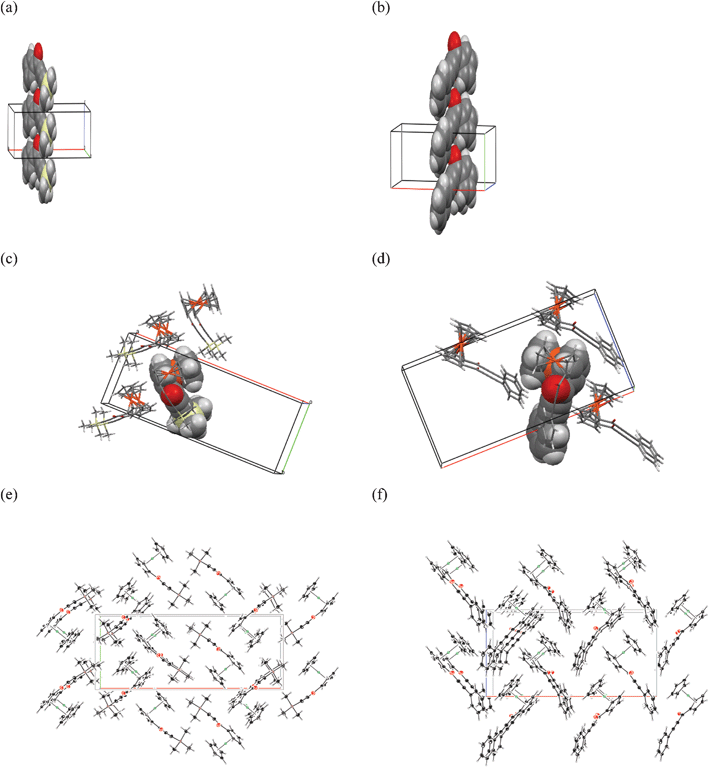 | ||
| Fig. 5 The similarities in crystal packing of 2c and 2d: the stacks of closely interacting molecules in space fill representation of 2c (a) and 2d (b), the more important intermolecular interactions of 2c (c) and 2d (d) and crystal packing diagrams in 001 direction for 2c (e) and in 010 direction for 2d (f). | ||
The interactions between the stacks are further determined by a character of the molecule and the accessibility of the carbonyl oxygen atom. In the case of 2d, the more flat and rigid substituent does not shield the carbonyl oxygen from the neighboring molecule completely, enabling the formation of additional C–H⋯O short contact from the aromatic phenyl C–H group belonging to a molecule of the adjacent layer. This interaction is accompanied by short C–H⋯Cp contacts. A very short H⋯H contact appears between the H(19) and H(17) atom from the adjacent stack related by a 2(1)[001] screw axis as a result of inter-stack interactions. The numerous short C–H⋯π interactions between the ferrocenyl moieties and the phenyl substituents of the molecules provide further means of close and effective packing.
The carbonyl oxygen in the case of 2c is more immersed in a cavity shielded by C(14) and C(15) methyl groups. Nevertheless, a short C–H⋯O contact can still be formed by molecules from one of the adjacent layer (Fig. 5d (left)). On the other side of the stack, the bulky TMS moiety has difficult access to the vicinity of the oxygen from a neighboring stack, but the hydrogens from C(16) methyl group are directed towards the plane of the unsubstituted Cp ring. The size of TMS and formation of methyl to methyl contacts in the lattice results in the case of 2c in slightly less dense packing and longer crystallographic x axis, than in the case of 2d, as it is presented in Fig. 5e and 5f.
In discussion of crystal structures of 2a–d it may of interest to remind that achiral molecules relatively rarely crystallize in noncentrosymmetric space groups (in the best case the chance is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4).18 It has been suggested that probability of crystallization in a noncentrosymmetric space group increases when the achiral molecule is rigid.19 Therefore, the observed tendency for crystallization in noncentrosymmetric space groups (3 compounds for 4) might be explained by a rigid nature of alkynoyl substituents.
:4).18 It has been suggested that probability of crystallization in a noncentrosymmetric space group increases when the achiral molecule is rigid.19 Therefore, the observed tendency for crystallization in noncentrosymmetric space groups (3 compounds for 4) might be explained by a rigid nature of alkynoyl substituents.
2.5. Quantum chemical calculations
The electronic structure and electronic transitions in 2a–d and 3 have been theoretically studied at the molecular level by means of density functional theory (DFT) and time-dependent DFT (TD-DFT) using the Gaussian 03W.E01 standard package and various functionals and basis functions. Comparison of calculated geometrical parameters with those measured by X-ray diffraction showed that the most accurate data were obtained using the B3PW91 functional with the 6-31G(d) basis set. For example, Table 6 shows the length of the Fe–C1 bond in 2d calculated by means of various methods, compared with the experimental (X-ray diffraction) value.| Method | Fe–C1 bond length (Å) |
|---|---|
| B3LYP/6-31G(d) | 2.04482 |
| B3PW91/6-31G(d) | 2.02255 |
| B3LYP/LANL2LB | 2.10070 |
| B3LYP/LANL2DZ | 2.10602 |
| LSDA/6-31G(d) | 1.97400 |
| X-ray diffraction | 2.0295(1) |
Fig. 6 shows the plot of the molecular frontier orbitals of complexes 2a–d and 3.
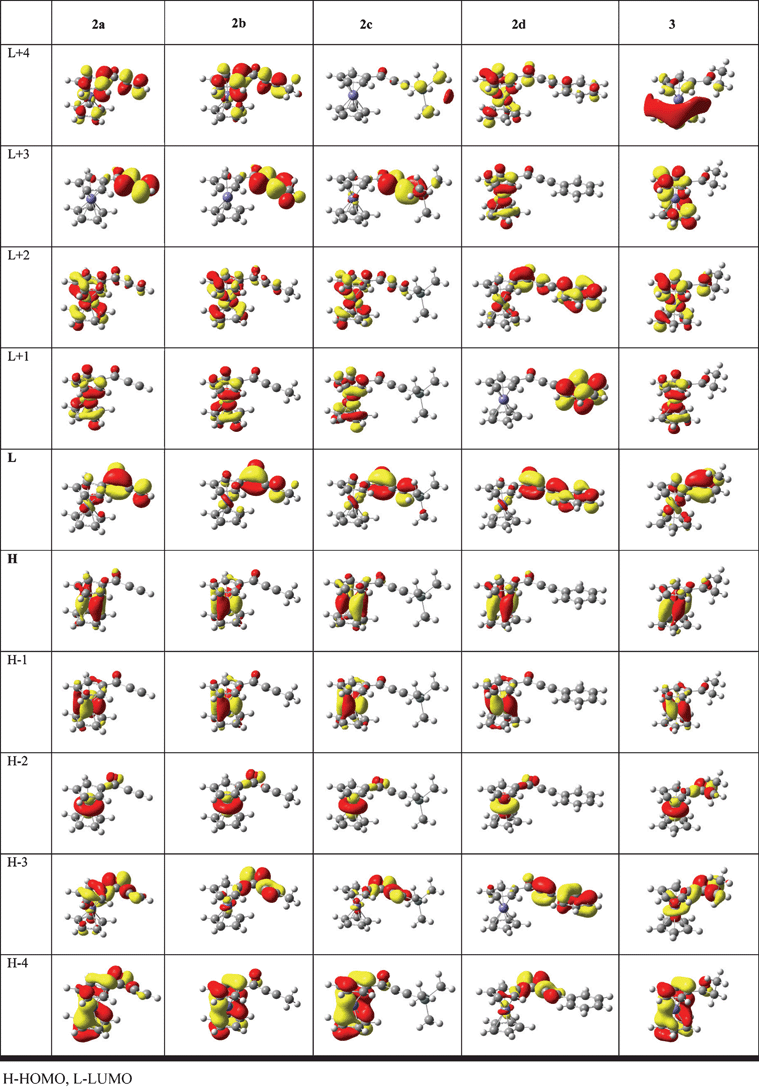 | ||
| Fig. 6 Molecular orbitals of 2a–d and 3 (contour value 0.04). | ||
The energies of these orbitals, calculated for isolated molecules and for chloroform solutions (using the polarizable continuum model, PCM) are listed in Table 7 and the Mulliken contributions of electrons localized on iron, Cp rings and lateral chain are presented in Table 8.
| Compound | Orbital energy for isolated molecules and (in parentheses) for CHCl3 solutions (eV) | ||||
|---|---|---|---|---|---|
| Orbitala | 2a | 2b | 2c | 2d | 3 |
| a H – HOMO, L – LUMO. | |||||
| L+3 | 0.953 | 1.142 | 0.379 | −0.038 | 1.577 |
| (0.919) | (1.651) | ||||
| L+2 | −0.090 | 0.017 | −0.056 | −0.490 | 0.152 |
| (−0.041) | (0.190) | ||||
| L+1 | −0.181 | −0.085 | −0.120 | −0.503 | −0.067 |
| (−0.118) | (−0.007) | ||||
| L | −1.806 | −1.563 | −1.779 | −2.027 | −0.990 |
| (−1.906) | (−1.075) | ||||
| H | −5.771 | −5.675 | −5.700 | −5.596 | −5.634 |
| (−5.713) | (−5.578) | ||||
| H-1 | −5.782 | −5.694 | −5.712 | −5.615 | −5.643 |
| (−5.724) | (−5.584) | ||||
| H-2 | −6.444 | −6.336 | −6.367 | −6.267 | −6.257 |
| (−6.387) | (−6.233) | ||||
| H-3 | −7.137 | −6.897 | −6.964 | −6.709 | −6.641 |
| (−7.107) | (−6.759) | ||||
| Orbital | Fe | C5H5 | C5H4 | RCO | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 2a | 2b | 2c | 2d | 3 | 2a | 2b | 2c | 2d | 3 | 2a | 2b | 2c | 2d | 3 | 2a | 2b | 2c | 2d | 3 | |
| LUMO+3 | 0.02 | 0.02 | 0.02 | 0.61 | 0.02 | 0.00 | 0.00 | 0.00 | 0.19 | 0.41 | 0.04 | 0.02 | 0.02 | 0.19 | 0.54 | 0.93 | 0.96 | 0.84 | 0.00 | 0.01 | |
| LUMO+2 | 0.55 | 0.54 | 0.55 | 0.21 | 0.45 | 0.17 | 0.16 | 0.17 | 0.06 | 0.14 | 0.20 | 0.21 | 0.20 | 0.08 | 0.30 | 0.07 | 0.08 | 0.09 | 0.64 | 0.09 | |
| LUMO+1 | 0.61 | 0.61 | 0.59 | 0.00 | 0.61 | 0.20 | 0.19 | 0.18 | 0.00 | 0.20 | 0.18 | 0.19 | 0.19 | 0.00 | 0.20 | 0.00 | 0.00 | 0.03 | 0.99 | 0.00 | |
| LUMO | 0.11 | 0.12 | 0.10 | 0.06 | 0.22 | 0.04 | 0.04 | 0.02 | 0.01 | 0.07 | 0.08 | 0.08 | 0.07 | 0.04 | 0.14 | 0.76 | 0.75 | 0.79 | 0.88 | 0.58 | |
| HOMO | 0.76 | 0.76 | 0.76 | 0.76 | 0.76 | 0.11 | 0.10 | 0.11 | 0.11 | 0.09 | 0.11 | 0.11 | 0.11 | 0.12 | 0.11 | 0.01 | 0.01 | 0.01 | 0.01 | 0.02 | |
| HOMO-1 | 0.78 | 0.78 | 0.78 | 0.78 | 0.78 | 0.10 | 0.11 | 0.10 | 0.10 | 0.10 | 0.11 | 0.11 | 0.11 | 0.10 | 0.10 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| HOMO-2 | 0.90 | 0.88 | 0.88 | 0.89 | 0.70 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 | 0.01 | 0.02 | 0.04 | 0.05 | 0.07 | 0.05 | 0.06 | 0.23 | |
| HOMO-3 | 0.04 | 0.06 | 0.05 | 0.01 | 0.25 | 0.09 | 0.00 | 0.00 | 0.03 | 0.00 | 0.23 | 0.07 | 0.09 | 0.04 | 0.10 | 0.62 | 0.83 | 0.85 | 0.91 | 0.64 | |
| HOMO-4 | 0.06 | 0.06 | 0.06 | 0.05 | 0.06 | 0.41 | 0.43 | 0.47 | 0.00 | 0.42 | 0.26 | 0.40 | 0.41 | 0.09 | 0.42 | 0.27 | 0.06 | 0.06 | 0.82 | 0.08 | |
As expected for the low-spin ferrocenyl compounds the HOMO, HOMO-1, and HOMO-2 orbitals are predominantly iron-centered dx2-y2, dxy and dz2 orbitals. The contribution of iron orbitals to these molecular orbitals is in the range of 70–90%. They are stabilized by the conjugation of the carbonyl group with the CC bond . The HOMO energy of 2a in chloroform is ∼0.14 eV lower than that of 3.
On the other hand, the LUMO's of 2a–d and 3 are predominantly localized at the lateral chains (75–88% and 56%, respectively) . Therefore, the HOMO-LUMO transitions for these compounds have a significant MLCT character. The LUMO energies decrease in the order 3 > 2b > 2c > 2a > 2d. The energy of LUMO of 2d is ∼1 eV lower than that of 3. Therefore, on going from the saturated ketone 3 to the ynones 2a–d we observe a weak stabilization of metal-centered HOMO orbitals and a much stronger stabilization of the lateral chain-centered LUMO orbitals. This brings about decrease of the HOMO-LUMO gap in the order 3 > 2b > 2a ≈ 2c > 2d. The LUMO+1 orbitals of are localized at the ferrocenyl moiety and result from interaction of metal d orbitals with the orbitals of the Cp rings. Interestingly, in the case of 2d analogous orbital is localized almost exclusively (99%) on the phenyl ring. A similar situation is observed for the LUMO+2 orbitals, which are predominantly localized on the iron atom and Cp rings for all compounds except 2d, for which this orbital is mainly centred (64%) on the lateral chain.
C bond increases the contribution of the HOMO-LUMO, HOMO-LUMO+1 and HOMO-LUMO+2 transitions to the LE band. As far as the HE bands are concerned the same effect is observed for the HOMO-LUMO, HOMO-1-LUMO+1 and HOMO-2-LUMO transitions.
| Compound | Calculated (CHCl3) nm (eV) | fa | Calculated (isolated molecule) nm (eV) | fa | Composition (CHCl3)% | Experimental nm (eV) |
|---|---|---|---|---|---|---|
| a oscillator strength. H—HOMO, L—LUMO. | ||||||
| 2a | 510 (2.43) | 0.0114 | 498 (2.49) | 0.0064 | 47 H→L | 492 (2.52) |
| 30 H→L+2 | ||||||
| 13 H→L+1 | ||||||
| 390 (3.18) | 0.0102 | 379 (3.27) | 0.0075 | 38 H-1→L+1 | 375 (3.31) | |
| 27 H→L | ||||||
| 16 H-2→L | ||||||
| 2b | 502 (2.47) | 0.0108 | 491 (2.52) | 0.0058 | 37 H→L | 482 (2.58) |
| 35 H→L+2 | ||||||
| 18 H-1→L+1 | ||||||
| 382 (3.25) | 0.0110 | 371 (3.34) | 0.0074 | 31 H-1→L+1 | 368 (3.37) | |
| 28 H→L | ||||||
| 19 H-2→L | ||||||
| 2c | 519 (2.39) | 0.0215 | 499 (2.48) | 0.0082 | 45 H→L | 493 (2.52) |
| 16 H→L+4 | ||||||
| 15 H→L+1 | ||||||
| 12 H-1→L+3 | ||||||
| 406 (3.05) | 0.0182 | 382 (3.25) | 0.0099 | 33 H→L | 377 (2.98) | |
| 32 H-1→L+3 | ||||||
| 12 H-2→L | ||||||
| 8 H→L+4 | ||||||
| 2d | 512 (2.42) | 0.0139 | 505 (2.46) | 0.0122 | 46 H→L | 497 (2.49) |
| 29 H-2→L+2 | ||||||
| 11 H-1→L+1 | ||||||
| 395 (3.14) | 0.0125 | 396 (3.13) | 0.0151 | 37 H-1→L-1 | 366 (3.39) | |
| 28 H→L | ||||||
| 15 H-2→L | ||||||
| 6 H→L+2 | ||||||
| 3 | 490 (2.53) | 0.0036 | 484 (2.56) | 0.0099 | 35 H→L | 454 (2.73) |
| 27 H→L+2 | ||||||
| 22 H-1→L+1 | ||||||
| 360 (3.44) | 0.0042 | 353 (3.51) | 0.0023 | 27 H-1→L+1 | 335 (3.70) | |
| 22 H-2→L | ||||||
| 18 H→L | ||||||
| 5 H-2→L+2 | ||||||
This means that both absorption bands observed for compounds under study have stronger MLCT character for ynones 2a–d than for 3.
Quadratic hyperpolarizabilities of 2a–d. We have also calculated static quadratic hyperpolarizability tensors for isolated molecules of 2a–d and 3. The values of components of β and total first hyperpolarizabilities βtot5c are shown in Table 10.
| Compound | β xxx (a.u.) | β xxy (a.u.) | β xyy (a.u.) | β yyy (a.u.) | β xxz (a.u.) | β xyz (a.u.) | β yyz (a.u.) | β xzz (a.u.) | β yzz (a.u.) | β zzz (a.u.) | β tot [1030 esu] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a 1 a.u. = 8.6393 × 10−33 esu.5c | |||||||||||
| 2a | −700.15 | −22.232 | −5.641 | 34.400 | 99.625 | 27.4904 | 0.95086 | 47.518 | 10.417 | −32.786 | 5.707 |
| 2b | 453.97 | −260.113 | −12.202 | −25.483 | 233.143 | 34.0276 | −2.9365 | −11.366 | 17.977 | −52.487 | 3.746 |
| 2c | −581.83 | −356.937 | −80.001 | −107.41 | −207.415 | −35.9449 | 45.6909 | −18.599 | 10.073 | −25.5701 | 5.942 |
| 2d | −492.840 | 873.464 | 118.287 | 37.604 | −217.100 | −27.802 | 39.103 | 19.258 | −10.1218 | 34.858 | 3.093 |
| 3 | −360.726 | −45.3378 | −15.7767 | 21.2526 | 61.7882 | −1.53748 | −8.6433 | 18.141 | 13.081 | −40.4593 | 3.130 |
| Identification code | 2a | 2b | 2d | 2c | |||
|---|---|---|---|---|---|---|---|
| Empirical formula | C13 H10 Fe O | C14 H12 Fe O | C19 H14 Fe O | C16 H18 Fe O Si | |||
| Formula weight | 238.06 | 252.09 | 314.15 | 310.24 | |||
| T/K | 90(2) | ||||||
| Wavelength [Å] | 0.71073 (Mo-Kα) | ||||||
| Crystal system, space group | Orthorhombic, P212121 | Monoclinic, P21/n | Orthorhombic, Pna2(1) | Orthorhombic, Pna2(1) | |||
| Unit cell dimensions [Å, °] | |||||||
| a, α | 8.9366(9) 90 | 5.8159(4) 90 | 21.5703(8) 90 | 25.3890(9) 90 | |||
| b, β | 9.5424(10) 90 | 18.8274(15) 92.450(2) | 5.9474(2) 90 | 10.1455(4) 90 | |||
| c, γ | 12.1196(14) 90 | 10.1737(8) 90 | 10.9774(4) 90 | 5.8173(2) 90 | |||
| Volume/Å3 | 1033.52(19) | 1112.98(15) | 1408.26(9) | 1498.44(9) | |||
| Z, Calculated density [mg m−3] | 4, 1.530 | 4, 1.504 | 4, 1.482 | 4, 1.375 | |||
| Absorption coefficient [mm−1] | 1.42 | 1.33 | 1.07 | 1.08 | |||
| F(000) | 488 | 520 | 648 | 648 | |||
| Crystal size/mm | 0.28 × 0.22 × 0.1 | 0.19 × 0.10 × 0.03 | 0.29 × 0.22 × 0.09 | 0.35 × 0.1 × 0.09 | |||
| θ range for data collection (°) | 2.72 to 28.28 | 2.16 to 27.50 | 2.65 to 27.50 | 1.60 to 28.37 | |||
| Diffractometer | Bruker Kappa APEXII Ultra | ||||||
| Limiting indices | 0 ≤ h ≤ 11, 0≤≤12, −16 ≤ l ≤ 16 | −7 ≤ h ≤ 7, −24≤≤24, −13 ≤ l ≤ 13 | −26 ≤ h ≤ 28, −7≤≤7, −9 ≤ l ≤ 14 | −33 ≤ h ≤ 33, −13≤≤11, −7 ≤ l ≤ 7 | |||
| Reflections collected/unique R(int) | 2561/2561 | 13021/2567 |
17233/2754 |
12979/3660 |
|||
| 0.0216 | 0.0332 | 0.0232 | 0.0311 | ||||
| Completeness to θ = 28.28° (%) | 100 | 100 | 99.8 | 100 | |||
| Absorption correction | Numerical | ||||||
| Max. and min. transmission | 0.931 and 0.798 | 0.831 and 0.748 | 0.967 and 0.759 | 0.997 and 0.832 | |||
| Structure solution | direct method | ||||||
| Refinement method | Full-matrix least-squares on F2 | ||||||
| H atom treatment | constrained | ||||||
| Data/restraints/parameters | 2561/0/136 | 2567/0/146 | 2754/1/190 | 3660/1/176 | |||
| Goodness-of-fit on F2 | 1.09 | 1.06 | 1.05 | 1.19 | |||
| Final R indices [I > 2σ(I)] | |||||||
| R 1 wR2 | 0.0195, 0.0456 | 0.0268, 0.0636 | 0.0177, 0.0414 | 0.0253, 0.0651 | |||
| R indices (all data) | |||||||
| R 1 wR2 | 0.0208, 0.0460 | 0.0335, 0.0671 | 0.0179, 0.0415 | 0.0290, 0.0768 | |||
| Absolute structure parameter | 0.010(13) | 0.012(10) | 0.185(14) | ||||
| Largest diff. peak and hole/e Å−3 | 0.274 and −0.162 | 0.482 and −0.256 | 0.246 and −0.266 | 0.416 and −0.398 | |||
As expected, the molecular second-order NLO responses of all complexes under study are weak. Moreover, the conjugation of the carbonyl group with the acetylenic bond does not seem to influence hyperpolarizability of ferrocenyl ketones. This means that the observed bulk second-order NLO responses of 2a–d are essentially due to favorable crystal packings.
3. Conclusion
We have found that conjugated ferrocenyl alkynones display remarkable tendency for crystallization in noncentrosymmetric space group to give SHG active crystals, which may be explained by a rigid structure of the alkynoyl substituent. The conjugation of the carbonyl group with the CC bond increases its electron withdrawing character and leads to stabilization of iron-centred HOMO-2 to HOMO-orbitals and lateral-chain centred LUMO orbitals. The HE and LE energy bands observed in the electronic absorption spectra of ferrocenyl ynones and their saturated counterpart, propionylferrocene have significant MLCT character and may contribute to NLO properties of these compounds.
4. Experimental
4.1. SHG measurements
The measurements of SHG were carried out by the Kurtz–Perry powder technique20,21 using a nanosecond Nd/YAG pulsed (10 Hz) laser operating at λ = 1064 nm. The outcoming Stokes-shifted radiation at 1907 nm, generated by Raman effect in a hydrogen cell (1 m long, 50 atm.) was used as the fundamental beam. The SHG signal was selected through a suitable interference filter, detected by a photomultiplier and recorded on a Tektronic TDS 620B oscilloscope. The recorded efficiencies were expressed versus that of powdered (50–125 μm) urea.4.2. Cyclic voltammetry
The cyclic voltammetry experiments were performed on a AUTOLAB (Eco Chimie BV) apparatus in a three electrode system, where the working electrode was a Pt disk (φ = 1.5 mm), the reference electrode was ferrocene electrode and the counter electrode was a cylindrical Pt gauze. The experiments were carried out under argon in 0.1 M solutions in dichloromethane containing Bu4NBF4 as supporting electrolyte.4.3. X-ray structure determination
A summary of crystallographic data collection and refinement parameters are collected in Table 11. The experimental data for all compounds have been indexed, integrated and scaled in the APEXII22 suite of Bruker (SAINT23 and SADABS24). The structures were solved using SHELXS25 software and refinement were conducted by SHELXL and WINGX26 packages.The refinements were based on F2 for all reflections except those with negative intensities. Weighted R factors wR and all goodness-of-fit S values were based on F2, whereas conventional R factors were based on the amplitudes, with F set to zero for negative F2. The F02 > 2σ(F02) criterion was applied only for R factors calculation was not relevant to the choice of reflections for the refinement. The R factors based on F2 are for both structures about twice as large as those based on F. Scattering factors were taken from Tables 4.2.6.8 and 6.1.1.4 from the International Crystallographic Tables Vol.C.27In the final least-squares full-matrix refinement all non-hydrogen atoms for both structures were refined with anisotropic thermal displacement parameters. Hydrogen atoms were first located directly from the Fourier map, but then refined as riding atoms with idealized geometry. Thermal displacement parameters for the H atoms were refined isotropically.
The molecular graphics were prepared using ORTEP for Windows program.28.
4.4. Computational details
Density functional theory (DFT) calculations were performed by using Gaussian 03W.E01 (Version 6.1)29 at DFT level by using the B3LYP, B3PW91 and LSDA functional and various basis functions (6-31G(d), LANL2LB, LANL2DZ). The geometry optimizations for compounds 2a,c,d and 3 were performed using B3PW91 functional and 6–31 g(d) basis function for C,H,O, and Fe atoms and 6–311++g(d,p) basis function for Si atom. Calculation in solution (chloroform) were performed using the Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM). The molecular orbitals (MO's) were calculated for each compounds at the optimized geometries. The MO's visualization were generated from checkpoint files using GaussView 3.07 (Isovalue 0.04). The time-dependent DFT (TD-DFT) studies were performed for singlet excited states (default in Gaussian) for 40 states. The Mulliken contributions of electrons were obtained using Chemissian software.30 The UV-Vis spectra were calculated using the SWizard program, revision 4.631,32 using the Gaussian model.References
- S. Barlow and S. R. Marder, Chem. Commun., 2000, 1555–1562 RSC.
- S. Barlow, H. H. Bunting, C. Ringham, J. C. Green, G. U. Bublitz, S. G. Boxer, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 1999, 121, 3715–3723 CrossRef CAS.
- T.L. Kinningbrugh, S. Salman, Y. A. Getmanenko, V. Coropceanu, W. W. Porter III, T. V. Timofeeva, A. J. Matzger, J.-L. Brédas, S. R. Marder and S. Barlow, Organometallics, 2009, 28, 1350–1357 CrossRef.
- Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.–Y. Jen, L. R. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758–2766 CrossRef CAS.
- (a) D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS; (b) D. R. Kanis, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 1992, 114, x-10357 CrossRef; (c) I. C. de Silva, R. M. De Silva and K. M. N. de Silva, J. Mol. Struct:THEOCHEM., 2005, 728, 141–145 CrossRef CAS.
- J. Heck, M. Dede, Ferrocene-Based Electro-Optical Materials in: P. Štěpnička (Ed.), Ferrocenes- Ligands Materials and Biomolecules, Wiley, Hoboken 2008, pp 319–392 (Chapter 9) Search PubMed.
- J. P. Morrall, G. T. Dalton, M. G. Humphrey and M. Samoc, Adv. Organometal. Chem., 2007, 55, 61–136 CrossRef.
- S. Di Bella, Chem. Soc. Rev., 2001, 30, 355–2006 RSC.
- B. J. Coe, R. J. Doherty, S. P. Foxon, E. C. Harper, M. Helliwell, J. Raftery, K. Clays, E. Frantz and B. S. Brunschwig, Organometallics, 2009, 28, 6880–6892 CrossRef CAS.
- (a) I. Janowska, J. Zakrzewski, K. Nakatani, M. Palusiak, M. Walak and H. Scholl, J. Organomet. Chem., 2006, 691, 323–330 CrossRef CAS; (b) I. Janowska, J. Zakrzewski, K. Nakatani, J. A. Delaire, M. Palusiak, M. Walak and H. Scholl, J. Organomet. Chem., 2003, 675, 35–41 CrossRef CAS.
- K. N. Jayaprakash, P. C. Ray, I. Matsuoka, M. M. Bhadhabe, V. G. Puranik, P. K. Das, H. Nishihara and A. Sarkar, Organometallics, 1999, 18, 3851–3858 CrossRef CAS.
- (a) V. Alain, M. Blanchard-Desce, C.- T. Chen, S. R. Marder, A. Fort and M. Barzoukas, Synth. Met., 1996, 81, 133–136 CrossRef CAS; (b) V. Alain, A. Fort, M. Barzoukas, C.- T. Chen, M. Blanchard-Desce, S. R. Marder and J. Perry, Inorg. Chim. Acta, 1996, 242, 43–49 CrossRef CAS.
- M. L. H. Green, S. R. Marder, M. E. Thompson, J. A. Bandy, D. Bloor, P. V. Kolinsky and R. J. Jones, Nature, 1987, 330, 360–362 CrossRef CAS.
- J. A. Mata, E. Peris, I. Asselberghs, R. Van Boxel and A. Persoons, New J. Chem., 2001, 25, 299–304 RSC.
- (a) G. G. A. Balavoine, J.-C. Daran, G. Iftime, P. G. Lacroix, E. Manoury, J. A. Delaire, I. Maltey-Fanton, K. Nakatani and S. Di Bella, Organometallics, 1999, 18, 21–29 CrossRef CAS; (b) J. Chiffre, F. Averseng, G. G. A. Balavoine, J.-C. Daran, G. Iftime, P. G. Lacroix, E. Manoury and K. Nakatani, Eur. J. Inorg. Chem., 2001, 2211–2226 Search PubMed.
- D. Plażuk and J. Zakrzewski, J. Organomet. Chem., 2009, 694, 1802–1806 CrossRef.
- Y. Yamaguchi, W. Ding, C. T. Sanderson, M. L. Border, M. J. Morgan and C. Kutal, Coord. Chem. Rev., 2007, 251, 512–524 CrossRef.
- S. M. Batterjee, M. I. Marzouk, M. E. Aazab and M. A. El-Hashash, Appl. Organomet. Chem., 2003, 17, 291–297 CrossRef CAS.
- E. Pidcock, Chem. Commun., 2005, 3457–3459 RSC.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798 CrossRef CAS.
- J. P. Dougherty and S. K. Kurtz, J. Appl. Crystallogr., 1976, 9, 145 CrossRef.
- APEXII-2008v1.0 Bruker Nonius 2007.
- SAINT V7.34A Bruker Nonius 2008.
- SADABS-2004/1 Bruker Nonius area detector scaling and absorption correction, 2008.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- G. X. Win and L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
- International Tables for Crystallography, Ed. A. J. C. Wilson, Kluwer: Dordrecht, 1992, Vol. C Search PubMed.
- ORTEP-3 for Windows: L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- Gaussian 03, Revision E.01-SMP, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 2003.
- http://www.chemissian.com/ .
- S. I. Gorelsky, SWizard program, http://www.sg-chem.net/.,University of Ottawa, Ottawa, Canada, 2010 Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
Footnote |
| † CCDC reference numbers 839178–839181. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra00037g |
| This journal is © The Royal Society of Chemistry 2012 |