Dramatic effect of modified boranes in diastereoselective reduction of chiral cyclic α-ketophosphinates†
Damien
Filippini
a,
Severine
Loiseau
a,
Norbert
Bakalara
b,
Zofia Anna
Dziuganowska
ac,
Arie
Van der Lee
d,
Jean-Noël
Volle
a,
David
Virieux
*a and
Jean-Luc
Pirat
*a
aAM2N, UMR 5253, ICGM, ENSCM, 8, rue de l'école normale, 34296 Montpellier cedex 5, France. E-mail: jean-luc.pirat@enscm.fr; david.virieux@enscm.fr; Fax: +33 4 6714 4319; Tel: +33 4 6714 7243
bINSERM U-1051, Hôpital Saint Eloi, rue Augustin Fliche, BP 74103, 34091, Montpellier, Cedex 5, France
cDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wroclaw University of Technology, Wroclaw, Poland
dInstitut Européen des membranes, cc047 Université de Montpellier 2, 34095, Montpellier, France
First published on 15th December 2011
Abstract
Phostines have been recently described as compounds having antiproliferative properties. Original synthesis of this new class of phosphinic analogs of pyranoses led to a mixture of four diastereomers 3–6 with unequal bioactivities. The most active compound 4 was originally obtained from a mixture of these four diastereomers by selective precipitation, giving firstly two diastereomers 3 and 4, epimers at the carbon atom. From the latter mixture 3 and 4, oxidation with Dess–Martin reagent afforded corresponding α-ketophosphinate 7, which by diastereoselective reduction using a chiral agent based on sodium borohydride and L-proline, gave preferentially the active diastereomer 4. In addition, use of a multivalent cation also increased the diastereoselectivity favourably.
α-Hydroxyphosphonates have received an increasing interest for the great diversity of biological functions illustrated by the inhibition of renin, EPSP synthetase, HIV protease or leucine aminopeptidase.1 Different studies have demonstrated that the absolute configuration of such α-position is decisive for the observation of the biological activity.2 In the exploration of new analogues of pyranosides containing a phosphorus atom, a new class of anticancer agents (i.e. “phostines”) recently emerged. This new family of phosphinolactones where the phosphorus atom is replacing the anomeric carbon, was specifically designed as a combination of a pyranoside and a C-aryglycoside. These compounds were synthesized from a two-step sequence, previously described by our group,3–5 by adding aryl-H-phosphinates 1 to protected arabinofuranose 2 in the presence of potassium tert-butoxide as catalyst. Four diastereomers (3–6), in 28/26/19/27 proportion respectively, were formed during this reaction arising from the creation of two stereocenters with moderate diastereoselectivies (Scheme 1). Interestingly the cytotoxic activities revealed them to be stereodependent and the glucose-like stereoisomer 4 was the most active one.4
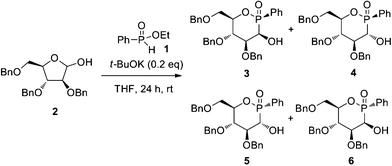 | ||
| Scheme 1 Synthesis of phosphinosugars. | ||
Unfortunately, the separation and the purification using preparative HPLC or recrystallization methods only gave the desired stereoisomer 4 in low yield. In this context, we explored a sequence involving an oxidation followed by a diastereoselective reduction of the epimers 3 and 4. The mixture of 3 and 4 was readily accessible by selective precipitation of the crude mixture of phostines 3–6 by addition of diethyl ether in 27% yield in a mixture of 3/4 (40/60).
To date, few publications deal with the oxidation of α-hydroxyphosphinates into α-ketophosphinates. A transposition from the phosphonate chemistry using Dess–Martin periodinane reagent (DMP) was evaluated. It was first reported by Ming Tao, DMP gave the corresponding α-keto-phosphonates or phosphinates in yields ranging from 30 to 92%.6 Thereafter, pyridinium dichromate was also tested by Mioskowski, leading to ketones in 60 to 80% yields.7
Oxidation was performed using these two procedures from the mixture of diastereomers 3 and 4 in the ratio 40/60. Dess–Martin oxidation was conducted in the presence of an excess of reagent at room temperature in dichloromethane (Scheme 2). The expected product 7 was effectively formed in 87% yield by 31P-NMR in CDCl3 giving one characteristic signal at 23.5 ppm confirming that 3 and 4 are epimers at the carbon atom. However, 7 was not isolated due to its sensitivity to air and moisture. Nevertheless, characterization of 7 was performed by mass spectroscopy analysis, as well as 13C-NMR spectroscopy which showed a representative doublet at 208.2 ppm (1JPC = 98.9 Hz), revealing the presence of the ketone function. The 1H NMR data also showed trans di-axial vicinal coupling constants confirming a chair structure for compound 7. Oxidation using pyridinium dichromate led to the desired product 7 along with many uncharacterized side-products.
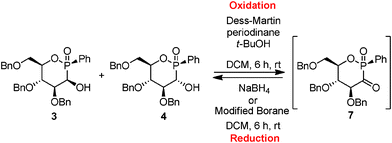 | ||
| Scheme 2 Oxidation of diastereomers 3 and 4 with Dess–Martin reagent and diastereoselective reduction of the α-ketophosphinate 7. | ||
Then, the enantiopure α-ketophosphinate 7 was used for diastereoselective reduction using modified boranes (Scheme 2). If ketophosphinate derivatives are an interesting class of activated electrophiles, no publication was reported for the diastereoselective reduction of this class of compounds and only a limited number of synthetic approaches was presented for enantioselective reduction of α-ketophosphonate into α-hydroxyphosphonate.8Oxidation and subsequently reduction were carried out as a “one pot” synthesis with only a final work-up (Table 1).
| Entry | Conditions (THF) | T °C | Time (h) |
dr
a (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) :4) |
de b | Yield (3:4) |
|---|---|---|---|---|---|---|
| a dr: diastereomeric ratio determined by 31P-NMR. b de: diastereomeric excess. | ||||||
| 1 | NaBH4 | rt | 5 | (67:33) |
34 | 86 |
| 2 | NaBH4 + (S,S)-tartaric acid | −30 | 48 | (79:21) |
58 | 94 |
| 3 | NaBH4 + (R,R)-tartaric acid | −30 | 48 | (82:18) |
64 | 49 |
| 4 | NaBH4 + CeCl3·7H2O | rt | 48 | (92:8) |
84 | 79 |
| 5 | NaBH4 + L-Proline | rt | 48 | (20:80) |
60 | 95 |
| 6 | NaBH4 + D-Proline | rt | 48 | (30:70) |
40 | 68.2 |
| 7 | NaBH4 + L-Proline + MgBr2.O(C2H5)2 | rt | 48 | (13:87) |
74 | 87 |
| 8 | NaBH4 + L-Proline + LiClO4 | rt | 48 | (10:90) |
80 | 76 |
| 9 | NaBH4 + L-Proline + ZnCl2 | rt | 48 | (11:89) |
78 | 78 |
| 10 | NaBH4 + L-Proline + CeCl3·7H2O | rt | 48 | (6:94) |
88 | 99 |
| 11 | NaBH4 + D-Proline + CeCl3·7H2O | rt | 48 | (10–90) | 80 | 84 |
Firstly, the reductions by sodium borohydride, and then by chiral boron-complex of (S,S)- or (R,R)-tartaric acids, and D- or L-prolines were tried. Sodium borohydride gave preferentially the undesired diastereomer 3 in 34% de (entry 1). Surprisingly, both (S,S)- or (R,R)-tartaric acid modified boranes also gave 3 in, respectively, 58% and 64% de (entry 2–3) showing almost low influence of the reagent stereochemistry. These preliminary results could be explained by the bulkiness of the reducing agents. The nucleophilic addition to cyclohexanone derivatives generally occurs by minimizing the 1,3-diaxial interactions. In such reactions, boranes attacked the Si face of the ketone leading to 3 (Fig. 1). Moreover stabilizing H-bonding association between the phosphoryl group (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the hydrogens of alcohol function of tartaric acid could also explain the increase of the diastereomeric ratio from 34% to 58% when sodium borohydride or (R,R)-tartaric acid were used. Nevertheless, a weak difference of de was noticed between (S,S)- and (R,R)-tartaric acid revealing a poor double stereo-differentiation between the mismatch pair with (S,S)-tartaric acid and the match one with (R,R)-tartaric acid. Luche reagent (entry 4) gave a higher diastereomeric excess of 84% in favour of 3. This result might be explained by a better complexation of the cerium salt by the carbonyl group leading to a compact transition state and then a better diastereomeric excess.
O) and the hydrogens of alcohol function of tartaric acid could also explain the increase of the diastereomeric ratio from 34% to 58% when sodium borohydride or (R,R)-tartaric acid were used. Nevertheless, a weak difference of de was noticed between (S,S)- and (R,R)-tartaric acid revealing a poor double stereo-differentiation between the mismatch pair with (S,S)-tartaric acid and the match one with (R,R)-tartaric acid. Luche reagent (entry 4) gave a higher diastereomeric excess of 84% in favour of 3. This result might be explained by a better complexation of the cerium salt by the carbonyl group leading to a compact transition state and then a better diastereomeric excess.
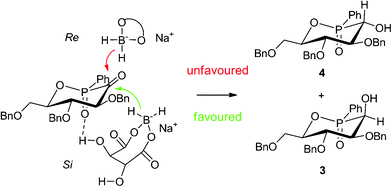 | ||
| Fig. 1 Approach of reductive complex of tartaric acid with sodium borohydride. | ||
Kolodiazhnyi reported that a complex of sodium borohydride with L-proline could invert the diastereoselectivity observed with tartaric acid.9 These conditions were tested and we were pleased to see an inversion of the stereoselectivity of the reaction in favor of the desired diastereomer (3/4 of 20:80, entry 5). Thereafter, a complex of sodium borohydride with D-proline was also used to check an eventual dependence of the chirality of the reducing agent. Nevertheless, the diastereomeric ratio remained almost unchanged showing a weak stereodifferentiation.
An explanation of these results would stem from the fact that proline deeply affects the nature of the interactions involved in the transition state. The reduction occurred on the Re face of the ketone due to the introduction of steric hindrance on the other side. It seemed possible to propose a complexation of the phosphoryl by a proline derivative leading to free the Re face of the keto-oxaphosphinane ring. These results clearly showed a strong diastereoselectivity in favour of 4, but which had to still be optimized.
Our efforts were oriented by Tanis's work, which reported the effect of inorganic cations on the reduction of ketones by borohydrides.10 This approach coupled to L-proline was attempted from our mixture of 3 and 4 with various bidente cation salts, such as magnesium bromide ethyl etherate, lithium perchlorate and zinc dichloride. The presence of complexing salt during the reduction process, increased dramatically the diastereomeric excess, and more particularly when using cerium trichloride, which reached 88% of de in favour of compound 4. Finally, all efforts to improve the diastereoselectivity, D-proline instead of L-proline, or by lowering temperature did not bring any conclusive improvement.
Conclusion
Diastereoselective reductions of chirally complex oxaphosphinanes was achieved by using cheap and readily affordable modified borane complexes. Tartaric acid complexes of borane led to the formation of the corresponding alcohol 3 by an attack to the less hindered face in the cis position regarding the phosphoryl group, which could be increased by using Luche reagent in higher diastereomeric excess of 88%. Whereas modified proline-borane gave selectively the other diastereomer 4. Addition of complexing cations increased the diastereoselectivity dramatically in the same way.Acknowledgements
Financial support from ANR Emergence Bio (IDEPHOST 2009-2011).References
- (a) D. V. Patel, K. Rielly-Gauvin and D. E. Ryono, Tetrahedron Lett., 1990, 31, 5587–5590 CrossRef CAS; (b) D. V. Patel, K. Rielly-Gauvin and D. E. Ryono, Tetrahedron Lett., 1990, 31, 5591–5594 CrossRef CAS; (c) J. A. Sikorski, M. J. Miller, D. S. Braccolini, D. G. Cleary, S. D. Corey, J. L. Font, K. J. Gruys, C. Y. Han, K. C. Lin, P. D. Pansegrau, J. E. Ream, D. Schnur, A. Shah and M. C. Walker, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 76, 115–118 CrossRef; (d) B. Stowasser, K. Budt, L. Jian-Qi, A. Peyman and D. Ruppert, Tetrahedron Lett., 1992, 33, 6625–6628 CrossRef CAS.
- (a) T. Kametami, K. Kigasawa, M. Hiragi, K. Wasisaka, S. Saga, K. Tanigawa, Y. Suzuki, K. Fukawa, O. Irino, O. Saita and S. Yambe, Heterocycles, 1981, 16, 1205–1211 CrossRef; (b) P. Kafarski and B. Lejczak, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 63, 193–215 CrossRef CAS.
- H.-J. Cristau, J. Monbrun, J. Schleiss, D. Virieux and J.-L. Pirat, Tetrahedron Lett., 2005, 46, 3741–3744 CrossRef CAS.
- J.-L. Pirat, D. Virieux, L. Clarion, J.-N. Volle, N. Bakalara, M. Mersel, J. Monbrun and H.-J. Cristau, PCT Int. Appl., 2009 Search PubMed WO 2009004096 A1 20090108.
- J.-N. Volle, D. Filippini, C. Midrier, D. Virieux, J.-L. Pirat, M. Sobecki and M. Drag, Synthesis, 2011, 2490–2494 CrossRef CAS.
- (a) M. Tao, R. Bihovsky, G. J. Wells and J. P. Mallamo, J. Med. Chem., 1998, 41, 3912–3916 CrossRef CAS; (b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- P. Vayron, P.-Y. Renard, A. Valleix and C. Mioskowski, Chem.–Eur. J., 2000, 6, 1050–1063 CrossRef CAS.
- (a) C. Meier and W. H. G. Laux, Tetrahedron: Asymmetry, 1995, 6, 1089–1092 CrossRef CAS; (b) C. Meier and W. H. G. Laux, Tetrahedron: Asymmetry, 1996, 7, 89–94 CrossRef CAS; (c) H. Gröger and B. Hammer, Chem.–Eur. J., 2000, 6, 943–98 CrossRef; (d) M. Bubenik, P. Préville, J. Dugas, G. Attardo and L. Chan, Tetrahedron Lett., 2003, 44, 8261–8263 CrossRef CAS.
- (a) C. Meier and W. H. G. Laux, Tetrahedron, 1996, 52, 589–598 CrossRef CAS; (b) A. Barco, S. Benetti, P. Bergamini, C. De Risi, P. Marchetti, G. P. Pollini and V. Zanirato, Tetrahedron Lett., 1999, 40, 7705–7708 CrossRef CAS; (c) V. V. Nesterov and O. I. Kolodyazhnyi, Russ. J. Gen. Chem., 2005, 75, 1161–1162 CrossRef CAS; (d) V. V. Nesterov and O. I. Kolodyazhnyi, Russ. J. Gen. Chem., 2006, 76, 1022–1030 CrossRef CAS; (e) V. V. Nesterov and O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2006, 17, 1023–1026 CrossRef CAS; (f) V. V. Nesterov and O. I. Kolodiazhnyi, Tetrahedron, 2007, 63, 6720–6731 CrossRef CAS; (g) I. Guliaiko, V. Nesterov, S. Sheiko, O. I. Kolodiazhnyi, M. Freytag, P. G. Jones and R. Schmutzler, Heteroat. Chem., 2008, 19, 133–139 CrossRef CAS.
- (a) S. P. Tanis, J. Org. Chem., 1988, 53, 4929–4938 CrossRef CAS; (b) M. B. Smith, Mc-Graw-Hill, Organic Synthesis, 1994, 380–382 Search PubMed; (c) C. E. Adams, F. J. Walker and K. B. Sharpless, J. Org. Chem., 1985, 50, 422–424 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra00799a |
| This journal is © The Royal Society of Chemistry 2012 |