A high throughput method for preparation of highly conductive functionalized graphene and conductive polymer nanocomposites†
Liping
Yang
,
Wu Aik
Yee
,
Si Lei
Phua
,
Junhua
Kong
,
Hui
Ding
,
Jun Wei
Cheah
and
Xuehong
Lu
*
School of Materials Science and Engineering, Nanyang Technological University, Singapore, 639798, Singapore. E-mail: asxhlu@ntu.edu.sg
First published on 27th January 2012
Abstract
Highly conductive graphene sheets were prepared by coating graphene oxide with polydopamine (PDA) followed by reduction with hydrazine. Polyacrylonitrile/graphene nanocomposites prepared via solution blending exhibit high electrical conductivities at very low graphene loadings owing to the good exfoliation and relatively planar conformation of the PDA-coated graphene in the polymer matrix.
Graphene-based polymer nanocomposites are promising materials for many applications, such as anodes in lithium ion batteries, semiconductors in electronic devices, electromagnetic shielding and shape memory devices.1 The development of graphene-based conductive nanocomposites requires graphene to be conveniently produced on a sufficiently large scale and dispersed in polymers as exfoliated sheets.2 The most promising method for large-scale production of graphene sheets is based on the exfoliation and reduction of graphene oxide (GO),1 and the reduced GO (RGO) can be dispersed into polymers as single-layer sheets via solution mixing.3 Using this method, the dispersion state of the graphene sheets in polymers is governed by the exfoliation level achieved prior to mixing.2 Since the RGO sheets tend to form agglomerates because of their decreased hydrophilicity after reduction,4 various approaches have been used to improve the dispersion of RGO, which include surface modification with surfactants and block polymers,5,6 phase transfer techniques,7in situ polymerization,8 grafting polymers onto RGO9 and directly mixing GO with polymers followed by reduction using hydrazine.2 These methods enabled the incorporation of graphene sheets into various polymers. However, most of these approaches are not suitable for large-scale preparation because of the difficulty to scale up, low yields or problems associated with processing-induced impurities.1,3
In this communication, we report a simple route for the preparation of water and organic-solvent dispersible RGO. It is based on a mussel-inspired biomimetic approach that is similar to the approach reported by the Messersmith group for bulk coatings; they used polydopamine (PDA), a component of mussel adhesion protein, as surface coatings on various organic and inorganic substrates to induce desirable surface adhesion properties.10 Recently, we developed a novel method to modify clay platelets, in which roughly a monolayer of PDA can be grown onto the surface of exfoliated clay platelets in water via controlling the polymerization time.11 It has also been reported that PDA can self-polymerize on the surface of GO and reduce the GO simultaneously to some extent.12 Unfortunately, the electrical conductivity of the as-prepared PDA-modified GO (DGO) is only two orders higher than that of the GO,12 making it unsuitable to be used as conductive fillers in polymers. However, it is worth noting that PDA is a semiconductor.13 If a monolayer of PDA is coated onto a properly reduced GO sheet, it may exhibit high electrical conductivity because of π–π stacking. Such properly reduced DGO sheets would also have a large amount of catechol groups on the surface and hence be highly hydrophilic and polar. We thus hypothesized that if DGO can be properly reduced using a simple chemical process, a high throughput route for the preparation of highly conductive graphene sheets that have good dispersibility in various solvents and polymer matrices may be realized.
We successfully reduced DGO in N,N-dimethylformamide (DMF) using a common reducing agent, hydrazine (Scheme 1). Prior to the reduction, dopamine polymerization time was set to be 4 h, which gave roughly a monolayer of PDA on the surface of GO.11 As no free PDA particles were formed in the suspension in the early stage of the polymerization, the DGO sheets could be easily extracted from the suspension and redispersed into DMF to carry out the reduction.
The thickness of the reduced DGO (RDGO) sheets were estimated via wide angle X-ray diffraction (WAXD) studies. The WAXD patterns of RDGO and reference samples (Fig. 1A) show that the characteristic diffraction peak of GO at 2θ = 10.8° (d-spacing 0.82 nm) is shifted to a smaller angle for DGO, and the peak becomes much broader. Considering that the DGO sheets are likely to be tightly stacked together owing to their surface catechol groups, the presence of a peak at 2θ = 7.6° for DGO implies that the thickness of a single DGO sheet is about 1.16 nm. The broadening indicates that the DGO stacks are more disordered than the GO stacks.11 In addition, a new broad peak corresponding to graphitic structure appears at 2θ = 24.5° (d-spacing 0.36 nm) for the DGO.12 It indicates that the GO is indeed reduced by PDA to some extent.12 After the DGO is further reduced by hydrazine, the peak at 2θ = 7.6° completely diminishes probably because of further disordering, and the broad peak ascribed to the graphitic structure becomes more intensive and sharper, indicating that most GO sheets have been reduced to graphene sheets. The thickness of the RDGO sheets was also investigated by atomic force microscopy (AFM) (Fig. S1†). The thicknesses of DGO and RDGO sheets are 1.25 nm and 1.26 nm, respectively, corroborating the WAXD data. The surface of the RDGO is smooth, indicating that a thin layer of PDA is evenly grown on the surface of the GO, and the hydrazine treatment did not destroy the structure of the PDA coating.
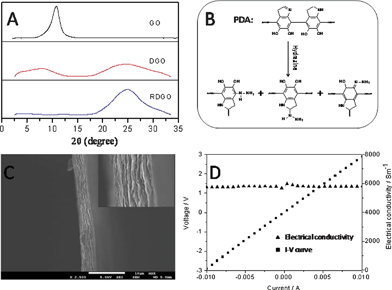 | ||
| Fig. 1 (A) WAXD patterns of GO (top), DGO (middle) and RDGO (bottom). (B) Possible chemical structure of PDA and hydrazine-treated PDA. (C) SEM image of a RDGO thin film with 4 μm thickness. (D) I–V curve of the RDGO thin film measured using the two-point probe method. | ||
X-ray photoelectron spectroscopy (XPS) was used to analyze the chemical structure of the RDGO. As shown in Fig. S2,† the XPS C 1s spectrum of GO can be divided into four components with binding energies at about 284.8, 286.9, 287.7 and 289.1 eV, corresponding to carbon atoms in C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO species, respectively.2 The XPS C 1s spectrum of DGO can be divided into five components with binding energies at about 284.6, 285.6, 286.6, 287.3 and 288.5 eV, respectively, which can be attributed to carbon atoms in C–C, C–N, C–O, CO and O–CO species, respectively. The appearance of the C–N peak at the binding energy of about 285.6 eV verifies the presence of PDA coating. The XPS C 1s spectrum of the RDGO can also be curve-fitted into the C–C, C–N, C–O, CO and O–CO species with binding energies at about 284.6, 285.5, 286.4, 287.8 and 288.7 eV, respectively. The intensities of the various components for GO, DGO and RDGO are listed in Table S1.† It shows that the PDA coating indeed reduces the GO to some extent, giving rise to a distinct decrease of C–O content. Hydrazine however further reduces the GO into a more perfect graphitic structure, leading to a remarkable decrease in CO and C–O content and a sharp increase in C–C content. Similar to other reported work,2 the O–CO species remains almost unchanged after the reduction. The hydrazine reduction process also causes an increase in C–N content, probably because some hydrazine molecules reacted with PDA layer via Michael addition reaction.10 Thus the surface of the RDGO sheets are likely to be rich in amine groups (Fig. 1B), which may help to increase the dispersibility of the RDGO sheets in various media. Raman spectroscopy provides additional evidence for the difference in the structures of the GO, DGO, and RDGO (Fig. S2†). They all exhibit D band at around 1337 cm−1 and G band at around 1566 cm−1. The DGO shows a similar D/G ratio (0.95) to that of the GO (0.94), while the RDGO shows a much higher D/G ratio (1.26), implying that more small domains of sp2 graphitic structure were further created upon hydrazine treatment.2 The TGA curves of the GO, DGO and RDGO are shown in Fig. S3.† Both the DGO and RDGO exhibit much higher thermal degradation temperatures than the GO.
O and O–CO species, respectively.2 The XPS C 1s spectrum of DGO can be divided into five components with binding energies at about 284.6, 285.6, 286.6, 287.3 and 288.5 eV, respectively, which can be attributed to carbon atoms in C–C, C–N, C–O, CO and O–CO species, respectively. The appearance of the C–N peak at the binding energy of about 285.6 eV verifies the presence of PDA coating. The XPS C 1s spectrum of the RDGO can also be curve-fitted into the C–C, C–N, C–O, CO and O–CO species with binding energies at about 284.6, 285.5, 286.4, 287.8 and 288.7 eV, respectively. The intensities of the various components for GO, DGO and RDGO are listed in Table S1.† It shows that the PDA coating indeed reduces the GO to some extent, giving rise to a distinct decrease of C–O content. Hydrazine however further reduces the GO into a more perfect graphitic structure, leading to a remarkable decrease in CO and C–O content and a sharp increase in C–C content. Similar to other reported work,2 the O–CO species remains almost unchanged after the reduction. The hydrazine reduction process also causes an increase in C–N content, probably because some hydrazine molecules reacted with PDA layer via Michael addition reaction.10 Thus the surface of the RDGO sheets are likely to be rich in amine groups (Fig. 1B), which may help to increase the dispersibility of the RDGO sheets in various media. Raman spectroscopy provides additional evidence for the difference in the structures of the GO, DGO, and RDGO (Fig. S2†). They all exhibit D band at around 1337 cm−1 and G band at around 1566 cm−1. The DGO shows a similar D/G ratio (0.95) to that of the GO (0.94), while the RDGO shows a much higher D/G ratio (1.26), implying that more small domains of sp2 graphitic structure were further created upon hydrazine treatment.2 The TGA curves of the GO, DGO and RDGO are shown in Fig. S3.† Both the DGO and RDGO exhibit much higher thermal degradation temperatures than the GO.
To further determine to what extent the reduction of the DGO sheets restores the electrical properties of the graphitic network, we measured the room-temperature electrical conductivities of solution-casted DGO and RDGO papers using two-point probe method. For the DGO paper, the conductivity is only about 0.25 S m−1, whereas for the RDGO paper, it is striking to see that it is increased by four orders to about 5800 S m−1 (Fig. 1D), which is close to that of pristine graphite, demonstrating the effectiveness of the reduction by hydrazine. Furthermore, the adhesive feature of the semi-conductive PDA coating reduces the contact resistance between the RDGO sheets, and the RDGO sheets exhibit fairly planar conformation (Fig. 1C) owing to the confinement imposed by the PDA coating; these may all contribute to the impressive high conductivity.14
The presence of a large amount of catechol, amine and imine groups on the surface of the RDGO sheets make them readily dispersible in various solvents, such as DMF, dimethyl sulfoxide(DMSO), formic acid, tetrahydrofuran (THF), acetone, ethanol, as shown in Fig. S4.† The RDGO suspensions in THF, acetone and ethanol are not stable; the sheets precipitate within several hours to several days. However, the RDGO suspensions in DMF, DMSO and formic acid are very stable; the suspensions are still homogeneous even after two months (Fig. 2).
 | ||
| Fig. 2 The dispersion states of the RDGO in various media after storage for 2 months: (A) DMF, (B) DMSO, (C) formic acid, (D) THF, (E) acetone, (F) ethanol, (G) 3% ammonium hydroxide solution, and (H) toluene. | ||
Furthermore, the RDGO can also be stably dispersed in water at a concentration around 0.05 mg ml−1. The dispersibility of the RDGO in water can be further improved by adding in a small amount of ammonium solution, ethylenediamine or DMF.15 The RDGO can be readily dispersed in solvents with high Hansen solubility parameters, but not in apolar solvents such as toluene.16
The RDGO sheets were incorporated into polyacrylonitrile (PAN) via solvent blending. From the scanning electron microscopy (SEM) image of the fracture surface of the PAN/RDGO nanocomposite with 0.67 vol% RDGO (Fig. 3A), we can observe some very thin sheets that are pulled out from the matrix, demonstrating that the RDGO sheets are very well exfoliated in the matrix. From the cross sectional images of the nanocomposites with 1.36 vol% (Fig. 3B) and 2.75 vol% RDGO (Fig. 3C), we can see that the graphene sheets are fairly flat, which are much less crumpled than the RGO obtained via physical routes and also relatively flatter than the RGO sheets prepared by direct reduction of GO with hydrazine.2 It is believed that the PDA coating may confine the conformation of the graphene sheets, which may benefit the electrical conductivity of the nanocomposites because of the high aspect ratio of the flattened graphene sheets.2 WAXD patterns of PAN and the nanocomposite with 2.75 vol% RDGO are shown in Fig. S5.† The two patterns show only slight difference, confirming the good dispersion of the RDGO in the nanocomposite.
 | ||
| Fig. 3 SEM images of the fracture surfaces of PAN/RDGO nanocomposites with RDGO loadings of (A) 0.67 vol%, (B) 1.36 vol% and (C) 2.75 vol%. | ||
The electrical conductivities of the PAN/RDGO nanocomposites are plotted as a function of RDGO volume content in Fig. 4. A sharp transition from insulating to conducting can be observed at a low percolation threshold. The electrical conductivity of the PAN/RDGO nanocomposite with 0.16 vol% RDGO is about 10−5 S m−1, which may be satisfactorily used as antistatic material. The electrical conductivity of the nanocomposite with 2 vol% RDGO is higher than 1 S m−1.
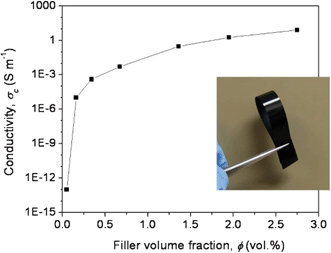 | ||
| Fig. 4 Electrical conductivity of the PAN/RDGO nanocomposites as a function of filler volume fraction. The inset is a digital photograph of the nanocomposite with 1.36 vol% RDGO. | ||
In summary, we developed a facile approach to prepare highly conductive functionalized graphene sheets that are dispersible in various media, and can be readily used to prepare conductive nanocomposites. The RDGO sheets can be easily prepared at gram scale using this high throughput method and it can be easily scaled up further. Moreover, the PDA coating also enhances the thermal stability and electrical conductivity of the RGO sheets effectively. Our approach is a generic route for preparation of stable suspensions of graphene sheets in various media and can be applied to prepare a wide variety of conductive polymer/graphene nanocomposites for different applications.
References
- (a) V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178 CrossRef CAS; (b) H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515 CrossRef CAS; (c) H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089 CrossRef CAS; (d) D. Y. Cai and M. Song, J. Mater. Chem., 2010, 20, 7906 RSC.
- (a) S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. Ruoff, Nature, 2006, 442, 282 CrossRef CAS; (b) T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327 CrossRef CAS; (c) S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155 RSC.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201 CrossRef CAS.
- X. Y. Qi, K. Y. Pu, H. Li, X. Z. Zhou, S. X. Wu, Q. L. Fan, B. Liu, F. Boey, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2010, 49, 9426 CrossRef CAS.
- (a) H. F. Yang, C. S. Shan, F. H. Li, Q. X. Zhang, D. X. Han and L. Niu, J. Mater. Chem., 2009, 19, 8856 RSC; (b) D. Vuluga, J. M. Thomassin, I. Molenberg, I. Huynen, B. Gilbert, C. Jerome, M. Alexandre and C. Detrembleur, Chem. Commun., 2011, 47, 2544 RSC.
- (a) P. G. Liu, K. C. Gong, P. Xiao and M. Xiao, J. Mater. Chem., 2000, 10, 933 RSC; (b) R. Verdejo, F. Barroso-Bujans, M. Angel Rodriguez-Perez, J. A. de Saja and M. Angel Lopez-Manchado, J. Mater. Chem., 2008, 18, 2221 RSC.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS.
- L. P. Yang, S. L. Phua, J. K. H. Teo, C. L. Toh, S. K. Lau, J. Ma and X. H. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 3026 CAS.
- (a) L. Q. Xu, W. J. Yang, K. G. Neoh, E. T. Kang and G. D. Fu, Macromolecules, 2010, 43, 8336 CrossRef CAS; (b) S. M. Kang, S. Park, D. Kim, S. Y. Park, R. S. Ruoff and H. Lee, Adv. Funct. Mater., 2010, 21, 108 CrossRef.
- J. McGinness, P. Corry and P. Proctor, Science, 1974, 183, 853 CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS.
- S. Park, J. H. An, I. W. Jung, R. D. Piner, S. J. An, X. S. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary characterisation data. See DOI: 10.1039/c2ra00798c |
| This journal is © The Royal Society of Chemistry 2012 |