Tailoring cobalt doped zinc oxide nanocrystals with high capacitance activity: factors affecting structure and surface morphology†
Marauo
Davis
,
Cenk
Gümeci
,
Bria
Black
,
Carol
Korzeniewski
and
Louisa
Hope-Weeks
*
Department of Chemistry and Biochemistry, Texas Tech University, Memorial Circle and Boston, Lubbock, TX 79409, USA. E-mail: Louisa.hope-weeks@ttu.edu; Fax: +18067421289; Tel: +18067424487
First published on 12th January 2012
Abstract
Highly crystalline zinc cobaltite (ZnCo2O4) nanocrystals were successfully synthesized through an epoxide driven, sol–gel method using Zn(NO3)·6H2O and CoCl2·6H2O as precursors. The crystal phase, morphology, specific surface areas, porosity, and capacitance activity of the prepared materials were characterized by powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), gas sorption techniques, and cyclic voltammetry, respectively. Results reveal that the synthesized nanocrystals are ∼4 nm in diameter. Electron microscopy studies illustrate significant changes brought on by varying the solvent and epoxide. Gas sorption analyses detail high specific surface areas (>200 m2 g−1) and porosities of the as prepared and annealed samples. Cyclic voltammetry experiments show that these zinc cobaltite nanocrystals have exceptional capacitance (∼700 Fg−1) and excellent cycle durability making them an excellent electrode material for supercapacitors.
Introduction
Supercapacitors are energy storage devices which have gained importance recently as potentially high power, alternative energy sources for use in electric vehicles and as memory backup for computers.1,2 Materials used in these devices typically have long cycle lives and should, for practical purposes, possess several intrinsic characteristics such as ease of preparation, low cost, and short charge–discharge times. In an effort to find new materials that exhibit enhanced electronic properties, conducting oxide aerogels have been investigated for their innate ability to amplify the nature of conducting oxides in batteries, ultracapacitors, and fuel cells.3 Typically, three kinds of electrode materials are used for fabrication of supercapacitors: high surface area carbon materials, such as carbon aerogels or nanotubes, conducting polymers, and transition metal oxides of Ru, Ni, Co, and Zn.4,5 Ruthenium oxides, for example, have illustrated the widest use and application in electrochemical supercapacitance studies.4 However, the high cost of Ru has limited its commercial prominence and has led to the study of other, cheaper metal oxides that exhibit variable oxidation states for supercapacitor applications.4 For example, nickel oxide6,7,8 cobalt oxide9 and manganese oxide materials are inexpensive and exhibit pseudocapacitive behavior similar to that of RuO2.4Spinel zinc cobaltite (ZnCo2O4) is a low-cost, binary metal oxide where Zn occupies tetrahedral sites in the structure and Co occupies the octahedral sites.10 In a recent study by the Renganathan group at Central Electrochemical Research Institute, found that ZnCo2O4 can, in fact, be prepared through coprecipitation.10 However, zinc cobaltite prepared from this method yielded low surface areas (<80 m2 g−1) with poor capacitance (∼70 Fg−1). Alternative approaches have been employed to produce materials of this class including wet-chemical methods such as sol–gel,11 and hydrothermal syntheses.12–14 In the present work, we report the synthesis and characterization of porous aerogel networks of ZnCo2O4 prepared via an epoxide driven, sol–gel method. To the best of our knowledge, this is the first report of the synthesis of binary zinc cobaltite aerogels from this method for application as a potentially viable supercapacitor. Optimal conditions of composition, crystallinity, specific surface area, pore volume, and pore size was achieved to yield zinc cobaltite aerogels of relatively high-specific capacitance.
The aim of this study is to investigate the synthesis and aerogel formation of zinc–cobalt mixed metal aerogels derived through the epoxide addition method. Both a stoichiometric (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and a non-stoichiometric (1:1) mixture of zinc nitrate and cobalt chloride were used as precursors for the preparation of zinc cobaltite. After gel formation, the materials were supercritically dried using liquid carbon dioxide. Following supercritical drying, the aerogels were calcined to yield the desired oxide phase, ZnCo2O4.
:2) and a non-stoichiometric (1:1) mixture of zinc nitrate and cobalt chloride were used as precursors for the preparation of zinc cobaltite. After gel formation, the materials were supercritically dried using liquid carbon dioxide. Following supercritical drying, the aerogels were calcined to yield the desired oxide phase, ZnCo2O4.
Experimental
Preparation of zinc cobaltite aerogels
Zinc nitrate hexahydrate, Zn(NO3)2·6H2O (Strem Chemicals, 98%), cobalt chloride hexahydrate, CoCl2·6H2O (Mallinckrodt Chemicals, 98%) were used as the precursors. The solvents: acetone (Mallinckrodt, 99.5%), UV Purified water, absolute ethanol (Parmco-Aaper, 200 Proof), 2-propanol (Sigma Aldrich), methanol (Mallinckrodt Chemicals), propylene oxide (ACROS Organics, 99.6%), n-butyl glycidyl ether (TCI America), and glycidol (ACROS Organics, 96%) were all used as received without purification. All syntheses were carried out under ambient conditions. The materials were prepared in both a stoichiometric (1:2) and a non-stoichiometric (1:1) molar ratio with respect to the desired ZnCo2O4 phase. All the results from the stoichiometric procedure can be found in electronic supplementary information.†
For stoichiometric preparation, Zn(NO3)2·6H2O (0.176 g, 0.60 mmol) was dissolved in methanol (1.25 mL) and stirred for approximately 2 min to give a colorless solution. Then to the resulting solution, CoCl2·6H2O (0.286 g, 1.20 mmol) in 2-propanol (2.50 mL) was added and then rapidly stirred for 2 min to yield a deep purple colored solution. Subsequently, propylene oxide was added (15 mmol) to the solution and stirred again for ∼20 s and then set aside to gel undisturbed for 24 h. Gels were obtained within 4 h.
In a typical synthesis for the non-stoichiometric ratio, preparation is as follows: Zn(NO3)2·6H2O (0.2230 g, 0.75 mmol) and CoCl2·6H2O (0.1784 g, 0.75 mmol) were dissolved in solvent (3.50 mL) and stirred for approximately 1 min to yield colored solutions of pink, blue, or purple (depending on solvent used). Such color changes are simply brought on by the difference in ligands at the metal center(s). Subsequently, epoxide (15 mmol) was added to the solution and stirred again for ∼20 s and then set aside to gel, undisturbed, for 24 h. Gels began to form within 3 h (see ESI Table 1†).
Following gelation, the gels were permitted to age ∼2 days to improve firmness and robustness. The resulting wet gels were then washed with acetone for ∼1 week with fresh acetone exchanged daily. Afterwards, the gels were processed in a SPI-DRY model critical point dryer. In the supercritical extractor, acetone was exchanged for liquid CO2 for 3–5 days, after which time the temperature was increased to 40 °C to achieve a pressure around 1300 psi. At this time, the dryer was slowly depressurized over a 4–6 h period to obtain monolithic aerogels.
To form the metal oxide, the aerogels were annealed in a programmable furnace to a temperature of 250 °C. The gels were heated from an initial temperature of 25 °C to the target of 250 °C at a rate of 1 °C min−1. Once at the target temperature, the samples were permitted to dwell at this temperature for 3 h before returning back to room temperature.
Physical characterizations
Powder XRD patterns of the samples were recorded with a Rigaku Ultima III diffractometer using Cu-Kα radiation. PXRD analyses were performed for both as prepared and annealed samples. To prepare the samples for X-ray powder diffraction, each sample was finely ground, and measurements were taken at a 2θ range of 10–80° at a step-width of 0.03° sec−1. Powder X-ray diffraction patterns were identified by comparison to the phases in the International Centre for Diffraction Data (ICDD) powder diffraction file (PDF) database.The surface morphology of the Zn–Co materials was studied using scanning electron microscopy (SEM) Hitachi S-4300. The aerogel powders were mounted on an aluminum stub with carbon tape. The mounted samples were then degassed at 100 °C for 24 h prior to imaging. The microscope was operated between 2 and 10 kV to minimize charging effects.
Transmission electron microscopy (TEM) analyses were conducted using a Hitachi H-8100 at an accelerating voltage of 150 kV while a JEOL JEM-2100, at an accelerating voltage 200 kV, was used for high-resolution TEM (HRTEM) images. The aerogel samples were prepared by lightly pressing the carbon-coated side of a copper grid onto the powder; excess powder was removed by passing a gentle stream of N2over the grid.
The specific surface area, pore volume, and average pore size of each material was obtained from the N2adsorption/desorption analyses conducted at 77 K on a Nova 4200e model Surface Area Analyzer (Quantachrome Instrument Corp.) The Brunauer-Emmett-Teller (BET) specific surface areas were calculated from the adsorption branch of the isotherm at 77 K. The pore size distributions, average pore diameters, and average pore volumes were all taken from the desorption branch employing the Barrett-Joyner-Halenda (BJH) model. Prior to obtaining any results, the samples were placed on degas for 24 h at 150 °C. Each set of measurements was taken at 77 mmHg and an equilibrium time of 600 s resulting in a total experimentation time of 6–9 h per sample.
Cyclic voltammetry measurements with a potentiostat (PC4/300, Gamry Instruments, Warminster, PA) were conducted to probe capacitive activity in a conventional three-electrode system in a 1M NaOH solution. A graphite carbon electrode with a 5 mm diameter served as the working electrode, while Ag/AgCl and a platinum wire were used as reference and counter electrodes, respectively. In a typical experiment, at least 0.50 mg cm−2 of the sample was loaded on the working electrode. The potential was scanned from 0.05 to 0.50 V to avoid the oxygen-evolution reaction with various scan rates.
Results and discussion
This work was intended to understand the factors that affect structure, surface morphology and capacitance activity of cobalt doped zinc oxide nanomaterials. No detailed characterization from the stoichiometric preparation is presented here but can be found in the electronic supplementary information.† Instead, we have specifically and deliberately discussed findings of the non-stoichiometric preparation allowing us to directly compare and contrast the effects of changing synthetic parameters. Also, it should be noted that upon aerogel formation of all the materials, the non-stoichiometric approach yielded more robust, stable monoliths with heightened physical properties and electrochemical activity as compared to those obtained from the stoichiometric approach.Gel formation studies
Cobalt doped zinc oxide gels were prepared using propylene oxide, glycidol, or n-butyl glycidyl ether as the gelation agent.The synthetic conditions and initial sol colors have been summarized and are shown in ESI Tables 1–3.† But, briefly, the materials prepared using propylene oxide will be labeled with “P” while those prepared using n-butyl glycidyl ether will be denoted by “B.” Likewise, “E” will denote if ethanol is used in preparation while “P” will signify 2-propanol.
Successful gel formation was observed with all solvents with the exception of water and methanol when propylene oxide was used as the gelation agent (see ESI Table 1†). The only significant differences that can be noted for the formed gels are gelation times, which varied depending on the solvent. For example, the shortest gel time is seen with 2-propanol (3–6 h) while the longest resulted from using acetone or ethanol (5–8 h). In the cases of no gel formation, the metal species quickly precipitated out and appeared as a suspended, “colloidal-like” system lacking robustness. This phenomenon may be explained by too fast an increase in solution pH resulting from the irreversible, ring opening of the highly strained epoxide. These precipitation reactions might also be attributed to the differences in solubility and stability of the colloids in solution.15
When using n-butyl glycidyl ether, gel formation is seen with only ethanol or 2-propanol. With this epoxide, the gelation time is noticeably longer than gels obtained when propylene oxide was used with corresponding solvents. From a theoretical viewpoint, these results can be explained simply by the slower reactivity of the “bulky” n-butyl glycidyl ether as compared to propylene oxide.
Finally, no stable gels could be formed with glycidol. Glycidol is different from the other epoxides since it is bifunctional (containing both epoxide and alcohol functionalities). For example, each solution prepared using glycidol, with the exception of acetone and water, resulted in highly unstable gels which lacked robustness even after considerable ageing, and, therefore, could not be dried to form an aerogel. Yet it can be assumed that since a “gel” did form, perhaps, investigating appropriate reaction conditions, stable gels should be achievable. However, to investigate this phenomenon was beyond the scope of this experiment.
Overall, since all metal precursor concentrations are constant and only solvent and epoxide is varied, we can correlate gelation time as a function of these parameters. Then comparing the gels from the same solvent, it can be deduced that the change in gelation rate is due simply to the reactivity of the epoxide used which is in agreement with initial findings of Gash et al.16 In theory, one might imagine a highly strained epoxide, like propylene oxide, for example, to exhibit a much faster irreversible, ring opening (i.e. increased reaction time) than n-butyl glycidyl ether. When the gelation time is longer, there is a slower and steadier increase in pH; and this permits more uniform assembly of the nanoparticles.
After gel formation and drying, all products appeared to be very monolithic and robust (see Fig. 1). From this point in the experiment, only materials PE, PP, BE and BP were used in analysis and further characterization since we could directly compare and contrast the effects introduced by choice of both solvent and epoxide.
 | ||
| Fig. 1 A) As prepared Zn–Co monoliths, PE and PP, prepared with n-butyl glycidyl ether in ethanol and 2-propanol, respectively. B) As prepared Zn–Co monoliths, BE and BP, prepared with propylene oxide in ethanol and 2-propanol, respectively. | ||
Powder X-ray diffraction
Aerogels prepared using both propylene oxide and n-butyl glycidyl ether were characterized by PXRD. Fig. 2 contains the powder X-ray diffraction patterns of the as prepared zinc–cobalt aerogels prepared with propylene oxide and n-butyl glycidyl ether, respectively. The diffraction patterns illustrate that all the samples are amphorous in nature with only weakly crystalline reflections. The weakly crystalline reflections are best assigned as resulting from Co(OH)2(PDF# 00-002-0925) and Zn(OH)2(PDF# 00-002-1437). Upon annealing at 250 °C, all samples were shown to become crystalline with major reflections resulting from the desired ZnCo2O4 (PDF# 00-023-1390) as shown in Fig. 3. Additionally, there are several other peaks corresponding to the formation of pure ZnO (PDF# 97-005-7156); which is consistent with the reaction stoichiometry. Even upon annealing to higher temperatures (350 °C and 450 °C), the materials remain stable, and the reflections become sharper and more clearly defined due to an increase in crystallite size (see ESI Fig. 1 and 2†).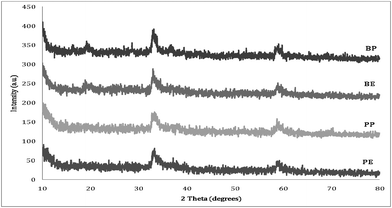 | ||
| Fig. 2 Powder X-ray diffraction patterns of as prepared zinc–cobalt aerogels prepared with propylene oxide (PE and PP) or n-butyl glycidyl ether (BE and BP). | ||
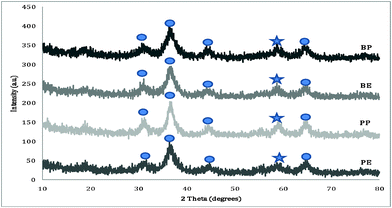 | ||
| Fig. 3 Powder X-ray diffraction patterns of 250 °C annealed zinc cobaltite aerogels prepared with propylene oxide (PE and PP) or n-butyl glycidyl ether (BE and BP). (ZnCo2O4is denoted with circles and ZnO with stars.). | ||
Electron microscopy
The surface morphology of the as prepared and annealed zinc–cobalt aerogels were evaluated using SEM. As shown in Fig. 4, in general, all as prepared samples exemplify a plate-like structure, and this surface morphology is retained upon annealing to 250 °C. This plate-like structure is typical of ZnO formation under similar conditions,17 and is consistent with the stoichiometric ratio and PXRD analysis. In further analysis of the SEM images, it is clear that the epoxide and solvent used directly results in subtle changes in morphology. For example, as shown in Fig. 4A and C, samples PE and BE, which are formed using ethanol as a solvent, exhibit a significantly larger platelet size and have a more feather like appearance compared to the materials prepared in 2-propanol which are shown in Fig. 4B and D. | ||
| Fig. 4 SEM images of Zn–Co aerogels: A–D) as prepared PE, PP, BE, and BP, respectively. E–H) annealed PE, PP, BE, and BP, respectively. | ||
The type of epoxide appears to have the greatest effect on morphology in the ethanol series where the BE sample exhibits a significantly larger platelet structure than the corresponding PE sample. It should, however, be noted that in changing the reaction solvent to 2-propanol, the effect of epoxide is minimized; as qualitatively, there is very little difference between samples PP and BP (see Fig. 4B and D). Such a finding indicates that there might be a coordination effect between reaction solvent and epoxide, and this conversely, results in the changing morphology.
Annealing of the aerogel materials, in most cases, results in a significant change of overall gel morphology. As shown in Fig. 4F and H, samples PP and BP undergo a very similar change in morphology, which exhibit a clearly defined platelet structure that appears to be more tightly aggregated when compared to the starting materials. The annealed BE sample shown in Fig. 4G appears to have become more closely packed while the average platelet particle size appears to have decreased, again, becoming more defined in appearance. These findings are supported by the N2 sorption measurements, which illustrate a larger surface induced by the decrease in particle size (see Table 1).
| Sample | Surface area [m2g−1] | Pore volume [cm3g−1] | Pore radius [nm] | Specific capacitance [Fg−1] |
|---|---|---|---|---|
| PE | 251(241) | 0.69(0.49) | 2.58(2.61) | 589(575) |
| PP | 203(176) | 0.44(0.28) | 2.60(2.60) | (452) |
| BE | 187(208) | 0.39(0.39) | 2.89(2.60) | (469) |
| BP | 151(187) | 0.45(0.42) | 2.60(2.60) | (482) |
The most dramatic change in gel morphology was observed upon annealing the PE sample, shown in Fig. 4E. Here, the aerogel appears to have undergone a transformation to result in a web-like structure composed of discrete particles that appear to have lost the defined platelet shape observed in all other samples. This change in morphology is in agreement with the N2 sorption analysis which shows that the PE sample exhibits a higher surface area than the other materials, due to the decrease in particle size. This decrease in particle size is also observed viaX-ray diffraction which illustrates that the average particle size is approximately 5 nm as indexed for the (311) reflection using the Scherrer's equation. This is in good agreement with the HRTEM analysis, shown in Fig. 5A, of the annealed PE aerogel sample, which shows that the material is composed of cubic particles that are ∼4 nm in size. As shown in Fig. 5B, the lattice d-space is calculated to be 0.23 nm indexed as (311) which correlates to the PXRD value of 0.24 nm for the (311) reflection calculated using the Bragg equation. Overall, it can be seen that slightly larger platelets result when ethanol is used as the solvent while propylene oxide, as the epoxide source, results in smaller particles.
 | ||
| Fig. 5 High-resolution TEM images of the PE aerogel annealed at 250 °C. | ||
Nitrogen adsorption/desorption analysis
Generally, the aerogel materials produced are structurally complex. As a result, Gash et al. has previously described erroneous pore size distribution and pore volume measurements from using this technique.16,18–21 However, increasing the equilibrium time during data collection is a reckoning for this issue and can eliminate the problem.21 Bearing this in mind, the listed results can be presented with a higher degree of accuracy. The surface area values by BET, pore volume and pore radii are illustrated for the as prepared zinc–cobalt aerogels as well as the calcined samples. Table 1 illustrates the obtained results from the N2 sorption analyses of the samples.As expected, the specific surface area values obtained for the as prepared materials were relatively high before being annealed. While comparing all materials, a higher surface area can be seen with PE (the highest) and PP samples as compared to BE and BP samples, respectively. The only difference in these materials is the gelation agent used; therefore it can be deduced that the epoxide directly affects the surface area in this case. Additionally, in comparing samples PE and PP with BE and BP, respectively, an obvious relationship relevant to solvent used can be identified. For example, PE and BE were prepared using ethanol while samples PP and BP used 2-propanol, and the higher surface areas result from the use of ethanol (see Fig. 4A and E). As a result, we can also deduce that solvent too directly affects surface area; this may also be supported by the idea that there is a higher dielectric constant (higher polarity) for ethanol as compared to 2-propanol which may better stabilize the species in the solution.
Once the materials were annealed, the surface area as well as the pore volume generally decreased. Such results are expected due to grain growth, upon annealing at a higher temperature.
Electrochemical measurements
Cyclic voltammetry (CV) measurements were conducted on all samples. However, more extensive measurements were performed on the PE sample to obtain stability and durability behavior.This sample was chosen for study due to its drastic changes in morphology and high surface area. Fig. 6A compares CVs of the as prepared and annealed PE aerogel samples. It should be noted that these CVs were obtained after 150 stabilization cycles.22 The capacitance values are tabulated in Table 1. The corresponding specific capacitance values were calculated by the negative sweep from 0.50 to 0.05 V.
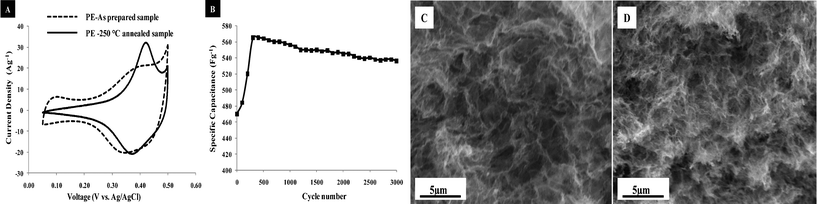 | ||
| Fig. 6 A) Cyclic voltammograms of as prepared and annealed sample in 1 M NaOH with a 25 mV s−1 scan rate, B) Stability test for the annealed PE sample. C and D) SEM micrographs of sample PE before and after electrochemical stability tests, respectively. | ||
Although the as prepared PE aerogel has a slightly higher capacitance (589 Fg−1) than the annealed PE aerogel sample (575 Fg−1), electrical double layer capacitance, which is more dominant in the as prepared sample, may account for this phenomenon. The greatest contributor to the response of the 250 °C, annealed sample is the pseudocapacitance arising from the redox chemistry of the sample. The pair of peaks near 0.40 V are associated with the Co(OH)2/CoOOH charge transfer processes. Moreover, it is clear that reversibility and sharpness of the CV curve is improved upon annealing indicating the high reversibility of the redox reaction of the annealed PE sample. The specific capacitance of this sample is calculated to be as high as 575 Fg−1 at a relatively high sweep rate (25 mV s−1), and this indicates its potential application for use as an electrode supercapacitor material. The obtained high specific capacitance of the annealed PE sample is higher than the PP, BE and BP annealed samples, (see Table 1) and this is possibly due to its very small particle size (∼4 nm), structurally interconnected porous morphology, and high specific surface area. The CV curve of the PE annealed sample is also much more symmetric than the PP annealed sample, therefore, illustrates higher redox reversibility (see ESI Fig. 3†). The specific capacitance value is also comparable to literature values for cobalt oxide5 and nickel cobaltite aerogels.23
Furthermore, as expected; specific capacitance values increase at low sweep rates. Additionally, it is observed that the specific capacitance value approaches 700 Fg−1 for the annealed PE sample at a 5 mV s−1 sweep rate (see ESI Fig. 4†).
Cycling stability and durability tests are critical parameters for the evaluation of a supercapacitor's use in long-term application. Fig. 6B reveals the cycling performance of the annealed PE sample. It was observed that nearly 95% of the specific capacitance was retained even after 3000 cycles, and that indicates the material’s outstanding durability. It is also very clear that structure morphology of the annealed PE sample is retained even after the 3000 cycles durability test (see Fig. 6C and D). We believe that ZnO, a well known cathodic inhibitor of corrosion, may play a key role in the exceptional durability of this material.
Conclusions
In summary, zinc cobaltite nanocrystals were successfully synthesized by a facile epoxide addition method with simple inorganic salts as raw materials. These prepared aerogel networks illustrated small particle sizes and relatively high specific surface areas that result in powerful supercapacitance and excellent durability. The internal structure and morphology can be tailored simply by varying solvent or epoxide. These factors were explored to achieve the optimal character of the materials. As a result, superior capacitance activity is illustrated through a series of electrochemical techniques. The findings here make this approach a viable, versatile process, and it further expands the repertoire for preparation of heterogeneous aerogel networks with useful application as promising electrode materials for supercapacitors.Acknowledgements
For the financial support provided by Texas Tech University's Provost Fellowship, the authors are exceedingly grateful. Also, a special thanks and appreciation must be extended to Sanjoy Bhattacharia and Grishma Khanal for all their assistance both in and out of the laboratory.References
- J. R. Miller and A. F. Burke, ECS Interface, 2008, 17, 53 CAS.
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539 CrossRef CAS.
- D. R. Rolison and B. Dunn, J. Mater. Chem., 2001, 11, 963 RSC.
- M. Selvakumar, D. Bhat, A. Aggarwal, S. Iyer and G. Sravani, Phys. B, 2010, 405, 2286 CrossRef CAS.
- T. Y. Wei, C. H. Chen, K. H. Chang, S. Y. Lu and C. C. Hu, Chem. Mater., 2009, 21, 3228 CrossRef CAS.
- K. C. Liu and M. A. Anderson, J. Electrochem. Soc., 1996, 143, 124 CrossRef CAS.
- C. N. Sisk and L. J. Hope-Weeks, J. Mater. Chem., 2008, 18, 2607 RSC.
- J. P. Zheng, P. J. Cygann and T. R. Zon, J. Electrochem. Soc., 1975, 142, 2699 CrossRef.
- V. Srinivasan and J. W. Weidner, J. Electrochem. Soc., 1997, 144, L210 CrossRef CAS.
- K. Karthikeyan, D. Kalpana and N. G. Renganathan, Ionics, 2009, 15, 107–110 CrossRef CAS.
- M. Ristic, S. Music, M. Ivanda and S. Popovic, J. Alloys Compd., 2005, 397, L1 CrossRef CAS.
- B. Baruwati, D. K. Kumar and S. V. Manorama, Sens. Actuators, B, 2006, 119, 676 CrossRef.
- Y. Chen, R. Z. Yu, Q. Shi, J. L. Qin and F. Zheng, Mater. Lett., 2007, 61, 4438 CrossRef CAS.
- A. E. Morales, M. H. Zaldivar and U. Pal, Opt. Mater., 2006, 29, 100 CrossRef.
- A. E. Gash, T. M. Tillotson, J. H. Jr. Satcher, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999 CrossRef CAS.
- A. E. Gash, J. H. Satcher Jr. and R. L. Simpson, Chem. Mater., 2003, 15, 3268 CrossRef CAS.
- Y. P. Gao, C. N. Sisk and L. J. Hope-Weeks, Chem. Mater., 2007, 19, 6007 CrossRef CAS.
- G. W. Scherer, J. Non-Cryst. Solids, 1998, 225, 192 CrossRef CAS.
- G. Reichenauer and G. W. Scherer, J. Non-Cryst. Solids, 2000, 277, 162 CrossRef CAS.
- G. Reichenauer and G. W. Scherer, J. Non-Cryst. Solids, 2001, 285, 167 CrossRef CAS.
- G. Reichenauer and G. W. Scherer, J. Colloid Interface Sci., 2000, 236, 385 CrossRef.
- C. C Hu, K. H. Chang and T. Y. Hsu, J. Electrochem. Soc., 2008, 185, F196 CrossRef.
- T. Y. Wei, C. H. Chen, H. C. Chien, S. Y. Lu and C. C. Hu, Adv. Mater., 2010, 22, 347 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic conditions, powder X-ray diffraction, electrochemistry measurements and all details on stoichoimetric procedure. See DOI: 10.1039/c2ra00793b |
| This journal is © The Royal Society of Chemistry 2012 |