Remarkable fluorescence enhancement in YVO4:Eu3+@Ag nano-hybrids induced by interface effect
Wen
Xu
,
Xue
Bai
*,
Sai
Xu
,
Yongsheng
Zhu
,
Lei
Xia
and
Hongwei
Song
*
State Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, 2699 Qianjin Street, Changchun, 130012, China. E-mail: hwsong2005@yahoo.com.cn; xuebai07@yahoo.com.cn; Fax: +86-431-85155129
First published on 12th January 2012
Abstract
The luminescence enhancement effect induced by the coupling of noble metal nanoparticles (NPs) with rare earth (RE) doped nanophosphors is attracting current interests. In order to obtain efficient luminescence enhancement and clarify its physical nature, the YVO4:Eu3+@Ag core-shell nano-hybrids colloids were first prepared by a facile two-step seed-mediated growth method. The luminescence enhancement of 5D0–7FJ transition for Eu3+ with a maximal factor of ∼one order was observed, which strongly depended on the amount of silver NPs, the concentration of the YVO4:Eu3+ colloids and the dissolved solutions. The enhancement mechanism was carefully discussed based on absorption spectra, emission spectra, luminescent decay dynamics and zeta potentials of colloids solutions. The results indicate that the silver NPs on the surface of YVO4:Eu3+ can effectively prevent VO43− and Eu3+ from the interaction with the solvent such as OH bonds, leading to a decrease of nonradiative energy transfer (ET) and suppressed luminescent quenching. A detailed model was proposed to explain the interesting luminescence enhancement behavior.
I. Introduction
Rare earth (RE) ions doped nanophosphors (NPs) are attracting extensive current attentions due to their potential applications in lasers,1 lighting and displays,2photon converters for solar cells,3 biological fluorescence imaging and detections,4etc. In contrast to the other phosphor species such as organic dyes and semiconductor quantum dots, RE doped NPs demonstrate unique photoluminescent properties, such as plentiful and narrow emission lines, long decay time constants, large stokes shift, quantum cutting and upconversion luminescence and so on.5–9 In addition, RE doped oxide and fluoride NPs demonstrate high chemical stability, high biocompatibility and low toxicity. These excellent properties make RE doped NPs an important representation in bio-applications.Although there has been much work in the field, it is still a challenge to effectively improve the brightness and luminescent quantum efficiency of RE doped NPs due to structure defects and their large surface areas with a variety of quenchers which have been formed in the previous preparation process.10,11 The brightness of RE doped NPs needs to be largely improved for further application, especially for the colloids. A well-known way to enhance the fluorescence of RE doped NPs is the coupling of emitters to metallic surfaces or particles.12 In the vicinity of metal structures, the electric field distribution is altered and as a result, the excitation field and the emitted radiation can be enhanced. Recently, a number of works have been performed to improve the photoluminescence of RE doped NPs through the coupling of RE ions to noble metallic surfaces or particles13,14 (silver and gold) in colloids, glasses, or thin films.12,15 Perriat and co-workers reported the luminescence enhancement of Gd2O3:Tb3+ NPs attached on gold nanorods (NRs) in a colloidal solution of diethylene glycol and observed a 37-fold enhancement factor,16 the mechanism was attributed to the resonant energy transfer (ET) between the gold NRs and the RE NPs. Note that in most of literatures, the resonant ET leads to fluorescence quenching of emitters.17 Some authors observed highly plasmon-enhanced UC emissions from NaYF4:Yb3+, Er3+ (Tm3+) @Au or NaYF4:Yb3+, Er3+ @Ag colloids and obtained enhancement factors of several fold up to ∼two orders.12a,15f,15g,18 In the literature, two main mechanisms for fluorescence enhancement have been proposed. One mechanism is the surface plasmon induced absorption enhancement of emitters due to the enhancement of the local electric field at the metal nanostructure surface. Another mechanism is the increase in the radiative decay rate of emitters due to the resonance of the emission with the surface plasmon of noble metals. It should be pointed out that in most of literatures, there was a lack of creditable evidence for the origin of fluorescence enhancement. For instance, no one practically observed the absorption enhancement of RE emitters, even in those highly luminescence-enhanced systems. Some authors observed improved decay rate of RE emitters, and others observed a suppressed decay rate.
We believe that in colloids, the RE doped NPs are not only affected by the field enhancement effect, but also influenced by an interface effect in the presence of silver or gold nanoparticles/nanoshells. In this work, YVO4:Eu3+ NPs colloids were prepared, which had strong absorption in the UV region and high photoluminescent efficiency in the red region.19 To obtain enhanced fluorescence, YVO4:Eu3+ @ Ag nano-hybrids colloids were prepared.20 In the hybrid colloids, it is exciting to observe an enhancement of an order of magnitude higher than the original. In addition, the interaction mechanism between emitters and the silver was systemically studied, which showed that the interface effect, solvent effect and concentration effect were of key importance for the luminescence enhancement of YVO4:Eu3+ @ Ag nano-hybrid colloids.
II. Experimental section
A. Synthesis of YVO4:Eu3+ spheres and YVO4:Eu3+ @ Ag Hybrids
The YVO4:Eu3+ spheres (∼150 nm) were prepared via the solvothermal method using DMF as the solvent, as according to our previous literature.21 In a typical synthesis, 0.25 g of Y(NO3)3·5H2O, 0.01 g of Eu(NO3)3·5H2O were first dissolved in DMF (10 ml), then a given amount of CTAB and PVP were added with stirring for one hour, and then 1 ml 2 mol l−1HCl aqueous solution and 0.22 g of Na3VO4·12H2O were subsequently added with stirring for another 12 h until the solution became homogenous. After that, the mixture was sealed in a Teflon-lined stainless steel autoclave and heated at 150 °C for 24 h under static conditions and then cooled naturally. YVO4:Eu3+ precipitates were collected and washed three times with alcohol, then air-dried at 60 °C for 24 h. All of the above-mentioned actions were performed under ambient atmosphere.The coating process was finished by the facile seed-mediated growth procedure,20 which was simply divided into two steps, seed attachment and seed growth, as shown in the Scheme 1. In the first step, the YVO4:Eu3+ powders (0.04 g) were dispersed in ethanol (10 ml). The suspension was added to the mixed solvents (12 ml, ethanol/acetone volume ratio = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) which was saturated with a certain amount of AgNO3 under magnetic stirring at room temperature for one hour. Then the colloids were washed with ethanol by centrifugation to remove excess silver ions, and dispersed in ethanol (20 ml) again. Subsequently, NaBH4 aqueous solution (0.1 M, 1 ml) was added. After one hour, the colloids were once more washed with ethanol to remove excess NaBH4 and dispersed in ethanol (40 ml) for standby. In the second step, a certain amount of standby colloid was added to aqueous and ethanol mixing solvents (volume ratio = 1:1) containing sodium nitrate (0.1 M), and different amount of silver nitrate. After that, the same amount of ammonia solution and L-ascorbic acid solution (0.1 M, 1 ml) was added. Finally, the composite colloids were centrifuged and dispersed in ethanol (5 ml) and the mass concentration was about 1 mg ml−1. The corresponding nano-hybrid powders were obtained by air-drying at 60 °C for 24 h.
:1) which was saturated with a certain amount of AgNO3 under magnetic stirring at room temperature for one hour. Then the colloids were washed with ethanol by centrifugation to remove excess silver ions, and dispersed in ethanol (20 ml) again. Subsequently, NaBH4 aqueous solution (0.1 M, 1 ml) was added. After one hour, the colloids were once more washed with ethanol to remove excess NaBH4 and dispersed in ethanol (40 ml) for standby. In the second step, a certain amount of standby colloid was added to aqueous and ethanol mixing solvents (volume ratio = 1:1) containing sodium nitrate (0.1 M), and different amount of silver nitrate. After that, the same amount of ammonia solution and L-ascorbic acid solution (0.1 M, 1 ml) was added. Finally, the composite colloids were centrifuged and dispersed in ethanol (5 ml) and the mass concentration was about 1 mg ml−1. The corresponding nano-hybrid powders were obtained by air-drying at 60 °C for 24 h.
 | ||
| Scheme 1 Schematic illustration of the formation of the YVO4:Eu3+@Ag nano-hybrids. | ||
B. Characterization
The surface morphology of the as-prepared products was recorded on a Hitachi H-8100 IV transmission electron microscope (TEM) under an acceleration voltage of 200 kV. The phase structure and purity of the as-prepared samples were characterized by X-ray power diffraction (XRD) with a Rigaku D/max 2550 X-ray diffractometer, using a monochromatized Cu target radiation resource (λ = 1.54 Å). The absorption spectra were measured with a UV-1800 UV-visible spectrometer. The excitation and emission spectra were recorded at room temperature using a Fluorescence Spectrophotometer. The luminescent quantum efficiency was measured with a Fluorescence spectrophotometer equipped with a BaSO4 integrated sphere. The luminescent dynamics were investigated by a laser-system consisting of a Nd:YAG pumping laser (1064-nm), the third-order Harmonic-Generator (355-nm) and a tunable optical parameter oscillator (OPO, Continuum Precision II 8000). It was with a pulse duration of 10 ns, repetition frequency of 10 Hz and line width of 4–7 cm−1. The zeta potential was investigated using a GG301-Nicomp 380/Zeta PLAS.III. Results and discussion
A. Morphology and structure of YVO4:Eu3+ @ Ag Nano-hybrids
First, the morphology of the YVO4:Eu3+ spheres and YVO4:Eu3+ @Ag hybrids coated with different amount of silver NPs were examined by HR-TEM images, as shown in Fig. 1. It can be seen that the YVO4: Eu3+ NPs before coating are smooth, uniform and mono-dispersed spheres, with an average diameter of ∼150 nm (see Fig. 1(a)). After first-step coating, the surface of YVO4: Eu3+ becomes rough and irregular, implying the formation of very small Ag NPs on the surface (see Fig. 1(b)). After second-step coating with different amounts of silver nitrate ∼5 and 200 μM, many distinguishable silver NPs (∼8 nm) adhere to the surface of YVO4:Eu3+ NPs, and the number of silver nanoparticles increases largely with the increase of silver nitrate. This structure is similar to some previous literature,12a,15g,18,22 on NaYF4:Yb3+, Er3+ @Au hybrids. | ||
| Fig. 1 TEM images of the YVO4:Eu3+ NPs (a) and YVO4:Eu3+@Ag hybrids prepared with different amount of silver nitrate in second-step deposition (b) 0 μM, (c) 5 μM, and (d) 200 μM. | ||
Fig. 2 shows the XRD patterns of the YVO4:Eu3+ NPs, YVO4:Eu3+@Ag hybrids and the corresponding standard cards, JCPDS 17-0341 for tetragonal YVO4 and 87-0717 for cubic silver. It can be seen that the YVO4:Eu3+ sample (curve a) is exactly in agreement with the corresponding standard cards, JCPDS 17-0341 for tetragonal YVO4. In the YVO4:Eu3+@Ag hybrids, the *311 face of cubic silver (marked with star) can be clearly distinguished. The *111, *200 and *220 faces for cubic silver overlap with the 202, 103, 204 faces of tetragonal YVO4, respectively. In the hybrids, the patterns corresponding to *111, *200 and *220 faces relatively increase in contrast to pure YVO4:Eu3+ (see curves b and c), further indicating the formation of YVO4:Eu3+@Ag hybrids.
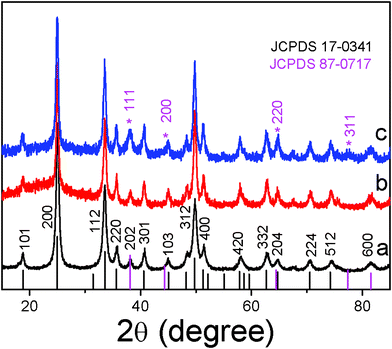 | ||
| Fig. 2 The XRD patterns of the YVO4:Eu3+ NPs (a) and YVO4:Eu3+@Ag hybrids prepared with different amount of silver nitrate (b) 5 μM, and (c) 200 μM. | ||
Fig. 3 shows the absorption spectra of YVO4:Eu3+ NPs and YVO4:Eu3+@Ag hybrids with different amounts of silver nitrate dispersed in ethanol solutions (The particle concentration was fixed at 0.07 mg ml−1). A band centering at ∼285 nm can be observed for all the solutions, corresponding to the charge transfer absorption from the oxygen to the central vanadium atoms inside the VO43−. The band location changes a little with the amount of silver nitrate because of unchanged energy gap between the ground and excited states of VO43− spaces.21 As the amount of silver nitrate increases to 200 μM, another peak appears at 450 nm, which could be assigned to the surface plasmon absorption of silver nanoparticles. The inset of Fig. 3 shows the absorbance at ∼285 nm as a function of silver nitrate amount. It can be seen that the absorbance of VO43−groups has little variation in the range of 0–16 μM and gradually decreases with the increase of silver nitrate in the range of 32–200 μM. This can be attributed to the scattering and reflection of visible and UV light by silver NPs adhered to the surface of the YVO4:Eu3+ NPs.
 | ||
| Fig. 3 Absorption spectra of YVO4:Eu3+ NPs and YVO4:Eu3+ @ Ag hybrids with various amounts of silver nitrate. | ||
B. Fluorescence enhancement of YVO4:Eu3+ @ Ag Hybrid Colloids
In YVO4:Eu3+@Ag hybrid colloids, a strong luminescence enhancement was observed—in contrast to the YVO4:Eu3+ NPs—which greatly dependent on the amount of silver NPs on the surface of YVO4:Eu3+ NPs and the concentration of YVO4:Eu3+@Ag in the colloids. Fig. 4(a) shows a comparison of emission spectra between 0.07 mg ml−1 of YVO4:Eu3+ NPs and YVO4:Eu3+@Ag (5 μM–AgNO3) hybrids in ethanol solution. The strong red emission of 5D0–7FJ (J = 0–4) for Eu3+ ions was distinguished and labelled in the figure. In contrast to YVO4:Eu3+ NPs, the overall fluorescence intensity of Eu3+ in the YVO4:Eu3+@Ag hybrid colloids increases up to 9.8 fold. Fig. 4(b) displays the fluorescence enhancement factor as a function of silver nitrate amount in YVO4:Eu3+@Ag hybrid colloids, which is defined as the ratio of the 5D0–7FJ emission intensity in YVO4:Eu3+@Ag hybrids to that in the YVO4:Eu3+ NPs. For the 0.07 mg ml−1 of YVO4:Eu3+@Ag solution, a 9.8 fold maximum enhancement factor was observed when the amount of silver nitrate was ∼5 μM. When the amount of silver nitrate increased continuously from 5 to 200 μM, the fluorescence enhancement factor gradually decreased, until it was 3.6 fold. For the 1.00 mg ml−1 of YVO4:Eu3+@Ag dense solution, the fluorescence enhancement factor also decreased with the increase in silver nitrate, varying from 1.6 to 1.15 fold. According to the inset of Fig. 3, it is suggested that the fluorescence suppression with the increase of silver nitrate in Fig. 4 is due to the decrease of incident light caused by the scattering and reflection of silver NPs.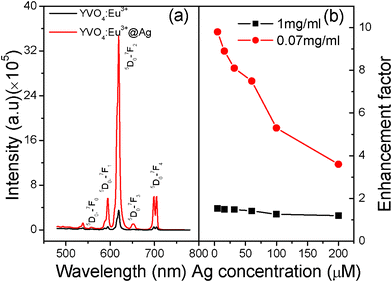 | ||
| Fig. 4 (a) A comparison of emission spectra between YVO4:Eu3+ NPs and YVO4:Eu3+ @Ag hybrids dispersed in ethanol solution (0.07 mg ml−1). (b) Luminescent enhancement factor as a function of silver nitrate (λex = 265 nm). | ||
The NP concentration in the YVO4:Eu3+ colloids has an obvious effect on the fluorescence intensity of both the YVO4:Eu3+ colloids23-25 and the YVO4:Eu3+@Ag nano-hybrids. Fig. 5 shows the dependence of the overall fluorescence intensity of Eu3+ on the particle concentration of YVO4:Eu3+or YVO4:Eu3+@ Ag colloids (5 μM silver nitrate). It is interesting to observe that the fluorescence intensity increases with the increasing particle concentration at first (0.01–0.07 mg ml−1) and approaches a maximum at 0.07 mg ml−1, before decreasing when the particle concentration exceeds 0.07 mg ml−1 for both the YVO4:Eu3+ NPs and the YVO4:Eu3+@Ag composites. This behavior indicates that if the concentration of NPs is too high, fluorescence quenching will be induced due to the interaction among different NPs.23 Despite this, the intensity of Eu3+ in the YVO4:Eu3+@Ag hybrids is significantly higher than for the YVO4:Eu3+ NPs. Similarly, the fluorescence enhancement factor is related to the particle concentration. It first increases from 6.0 to 9.8 when the concentration ranges between 0.01–0.07 mg ml−1, and approaches a maximum at 0.07 mg ml−1, then decreases from 9.8 to 1.6 as the concentration ranges between 0.07–1.00 mg ml−1.
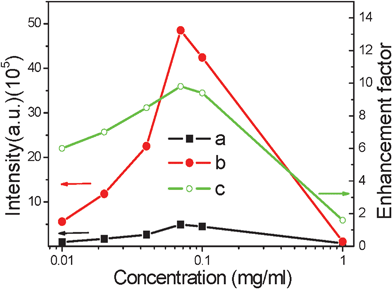 | ||
| Fig. 5 Dependence of luminescent intensity of 5D0–Σ7FJ of YVO4:Eu3+ (a) and YVO4:Eu3+ @ Ag hybrids (b) and the enhancement factor (c) on the particle concentration of YVO4:Eu3+ colloids (λex = 265 nm). | ||
The luminescent enhancement of YVO4:Eu3+@Ag colloids also strongly depends on the dissolved solutions. Table 1 lists the variation of luminescence enhancement factor with different solutions. It can be observed that as 0.01 mg ml−1 of the YVO4:Eu3+@Ag hybrids were dissolved in different solutions, DMF, EG, H2O and Ethanol, the emission intensity of Eu3+ increases 1.9, 2.9, 4.0 and 6.0 fold, in contrast to the corresponding YVO4:Eu3+ solutions. However, the intensity of Eu3+ in YVO4:Eu3+@Ag powders keeps nearly constant in comparison to the corresponding YVO4:Eu3+ powders. These results definitely suggest that the luminescence enhancement is induced by the interface effect between the solutions and the NPs, rather than the field enhancement effect, which will be discussed carefully in the next section. It is believed that the species, structure, polarity and optical properties of both solutions have strong influence on the luminescence enhancement of RE-doped NPs, which is a complex proposition to investigate.
C. Improved decay time constants in the YVO4:Eu3+ @ Ag Nano-hybrids
The luminescent decay dynamics of the 5D0–7F2 transition for YVO4:Eu3+@Ag nano-hybrids as well as YVO4:Eu3+ NPs were also studied. The results demonstrate that all the decay curves can be well fitted by a biexponential function, | (1) |
 | (2) |
 | ||
| Fig. 6 Dependence of decay time constant of 5D0–7F2 on the amount of silver nitrate (λex = 265 nm) in YVO4:Eu3+ NPs and YVO4:Eu3+ @ Ag hybrids. | ||
Actually, the particle concentration of YVO4:Eu3+ or YVO4:Eu3+@Ag also has an obvious influence on the decay time constant of 5D0→7F2 for Eu3+ ions. Fig. 7 shows the dependence of the decay time constants of the 5D0→7F2 transition on the particle concentration of YVO4:Eu3+ NPs or YVO4:Eu3+@Ag hybrids (5 μM silver nitrate). It is observed that as the particle concentration increases over a range of 0.01–1 mg ml−1, where the decay time constant inYVO4:Eu3+@Ag gradually increases from 127 to 260 μs. In the YVO4:Eu3+ NP colloids, the decay time constant has little variation when the concentration varies over 0.01–0.07 mg ml−1. And as the concentration changes from 0.07–1 mg ml−1, the decay time constant increases gradually from 74 to 141 μs. It is suggested that the increase of decay time constant with the increase of particle concentration is due to improvement of the reabsorption process. The reabsorption process represents the fraction of photons of the Eu3+ ions emission being absorbed once more, then remitted, reabsorbed, and so on. As a result, the decay time constant τ1 is prolonged, which can be approximately described as:26
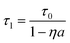 | (3) |
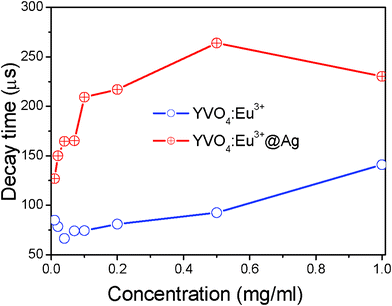 | ||
| Fig. 7 Dependence of the decay time constant of 5D0–7F2 on the particle concentration of YVO4:Eu3+ NPs or YVO4:Eu3+@Ag hybrids in solution (λex = 265 nm). | ||
The luminescent decay time constants of the 5D0→7F2 transitions between the YVO4:Eu3+ colloids and YVO4:Eu3+@ Ag hybrids in different solvents and powders were further compared, as shown in Table 1 It can be concluded that the decay time constants of YVO4:Eu3+@ Ag hybrid colloids become much longer than those in YVO4:Eu3+ NPs. However, the decay time constants of YVO4:Eu3+@ Ag hybrids powders in air become a little bit shorter than those in YVO4:Eu3+ NPs. In the powder samples, the nonradiative transition rates from 5D0 to the down levels 7FJ can be ignored due to very large energy gap of 5D0–7FJ (∼11200 cm−1) and smaller phonon energy (<1000 cm−1) of YVO4 lattices according to the multi-phonon relaxation theory.27 Therefore, the decay time constant of 5D0–7FJ transition is governed by the radiative transition rate. Because the radiative transition rate equals the inverse of the decay time constant, the radiative transition rate of 5D0–7FJ in YVO4:Eu3+@Ag hybrids increases a little in contrast to that in YVO4:Eu3+ NPs. This may be due to the variation of effective refractive index (neff) in different samples. In nanocrystalline YVO4:Eu3+ powders, when the particle size is less than 1/4 wavelength of emission light, the effective refractive index can be written as, neff ≈ xnYVO4++(1 − x)nair.28 In YVO4:Eu3+@Ag hybrids, neff ≈ xnYVO4++(1 − x)nAg. Because nAg > nair, the neff of YVO4:Eu3+@Ag should be bigger than that of YVO4:Eu3+. This will lead to the increase of radiative transition rate of 5D0–7FJ. Anyway, due to the low variation of the radiative transition rate, it can be concluded that the increase of the radiative decay rate of emitters is not the main reason for the luminescent enhancement.
D. The origin of luminescence enhancement behavior
Now let us carefully discuss the luminescent enhancement effect in YVO4:Eu3+@Ag hybrid colloids in comparison to the YVO4:Eu3+ NPs. According to the experimental results above, we consider that the main mechanism in this work for luminescent enhancement is neither the surface plasmon induced absorption enhancement of emitters due to the enhancement of the local electric field at the metal nanostructure surface, nor the increase of radiative decay rate of emitters due to the resonance of the emission with the surface plasmon of noble metals. The following facts are conflicted with the surface plasmon induced enhancement of the local electric field: (1) The absorption of VO43−groups decreases with the increasing amount of silver NPs, instead of increasing; (2) The luminescence enhancement was observed only in YVO4:Eu3+@Ag colloids, but not in the powders; (3) In YVO4:Eu3+@Ag hybrid colloids, the decay time constants of 5D0–7F2 increase greatly with the increased amount of silver NPs, but not in the powders, which should be independent of the enhancement of local electric field. The enhancement of the local electric field means an increase in incident light reaching the YVO4:Eu3+ NPs. We studied the dependence of the decay time constant of 5D0–Σ7FJ(J = 2) on the power density of the excitation light, and the result indicated that as the density of excitation power increased ∼one order, the decay time constant of 5D0–Σ7FJ(J = 2) rarely changed for both the YVO4:Eu3+ NPs and the YVO4:Eu3+@Ag hybrids, implying that the enhancement of the local electric field can not induce the variation of decay time constant. The luminescent enhancement in the present case is also inverse to the increase of radiative decay rate of emitters due to the resonance of the emission with the surface plasmon of silver, as described above. In addition, the emission range of 5D0–7FJ for Eu3+ ions (550–700 nm) is far away from the surface plasmon absorption of silver NPs and the luminescent enhancement in YVO4:Eu3+@Ag hybrid colloids is nearly independent of emission wavelength, as can be seen in Fig. 4a.Based on the above analysis, we believe that the main mechanism for the luminescent enhancement in YVO4:Eu3+@Ag hybrid colloids was induced by the interface effect in the solutions. To confirm this point, the quantum efficiency (QE) of YVO4:Eu3+@Ag hybrid colloids as a function of the silver nitrate amount was measured. The results demonstrated that the QE of pure YVO4:Eu3+ was only ∼0.3%. The QE of YVO4:Eu3+@Ag hybrid colloids was improved to ∼2.8% and nearly independent of the amount of silver nitrate as it varied from 5 μM to 100 μM. The QE increases about ∼9.3 fold after coating, which is close to the luminescent enhancement factor (∼9.8 fold). For the YVO4:Eu3+ and YVO4:Eu3+@ Ag nanopowders, the QE was determined to be ∼11.8%. This indicates that in the solutions, the QE of YVO4:Eu3+ decreases more significantly than the corresponding nanosized powders due to the interaction between YVO4:Eu3+ nanophosphors and the solutions. The formation of Ag NPs on the surface of YVO4:Eu3+ can restrict the interaction of nanophosphors and the solutions. Generally speaking, in RE doped NPs the involvement of surface large vibration bonds such as OH− , CO32− will induce PL quenching due to the increase of nonradiative ET rate from hosts, RE ions to these bonds.29 In the colloid solutions, the NPs should strongly interact with the solutions, such as ethanol, EG, DMF and H2O. In other words, the energy on excited states of the host lattices and RE ions could be transferred to solutions through the interface interaction of the NPs with large vibration bonds of solutions as they are within the range of effective interaction length. It is suggested that in the YVO4:Eu3+@Ag hybrid colloids, the interaction of NPs with large vibration bonds of solutions was hindered, thus the luminescence intensity and the decay time constant of the 5D0–7FJ transition increased due to the decrease of the nonradiative transition rate. In order to reveal how the interaction of NPs with large vibration bonds of solutions was hindered in the YVO4:Eu3+@Ag hybrid colloids, the zeta potentials of the YVO4:Eu3+ colloids in the seed-mediated growth process (First Step) and the YVO4:Eu3+@Ag hybrids (Second Step) were investigated, as shown in Table 2. It can be seen that the zeta potential of the original YVO4:Eu3+ colloids solution is positive. After the formation of YVO4:Eu3+@Ag hybrids, the zeta potential of the solution becomes negative. This means that the surface of YVO4:Eu3+ is charge-positive, while the YVO4:Eu3+@Ag hybrids are charge-negative. It is suggested that in the former case, YVO4:Eu3+ can be bound effectively with OH− bonds due to the charge gravitation between charge-positive NPs and charge-negative OH− bonds, while in the latter case the charge-negative YVO4:Eu3+@Ag and OH− bonds are repellant and can not be bound effectively each other. In the former case, ET from the surface of the YVO4:Eu3+NPs to OH− bonds happens easily, leading to the quenching of PL. On the contrary, ET from the surface of the YVO4:Eu3+@Ag hybrids to OH− bonds is suppressed, leading to luminescent enhancement. Fig. 8 gives the schematic illustration of the interface ET and PL processes of the YVO4:Eu3+ NPs and YVO4:Eu3+ @ Ag hybrids and the interaction mechanism for the YVO4:Eu3+ NPs and YVO4:Eu3+@Ag hybrids with ethanol solutions. In Fig. 8, when the VO43− species are excited, the energy will be transferred to the Eu3+ ions and the hydroxyl in the solvent, respectively and part of the energy transferred to the Eu3+ ions will further transfer to the hydroxyl in the solvent from the 5D0 level. Assuming that the radiative transition rate has little variation before and after silver coating, it is easy to deduce that the inner QE of Eu3+ on 5D0 is inverse to the lifetime of 5D0–7FJ. Then it can be estimated that the inner QE of Eu3+ in the YVO4 was improved ∼2 times, while the ET efficiency from VO43− species to Eu3+ ions was improved ∼5 times in the optimum conditions of the dissolved solutions.
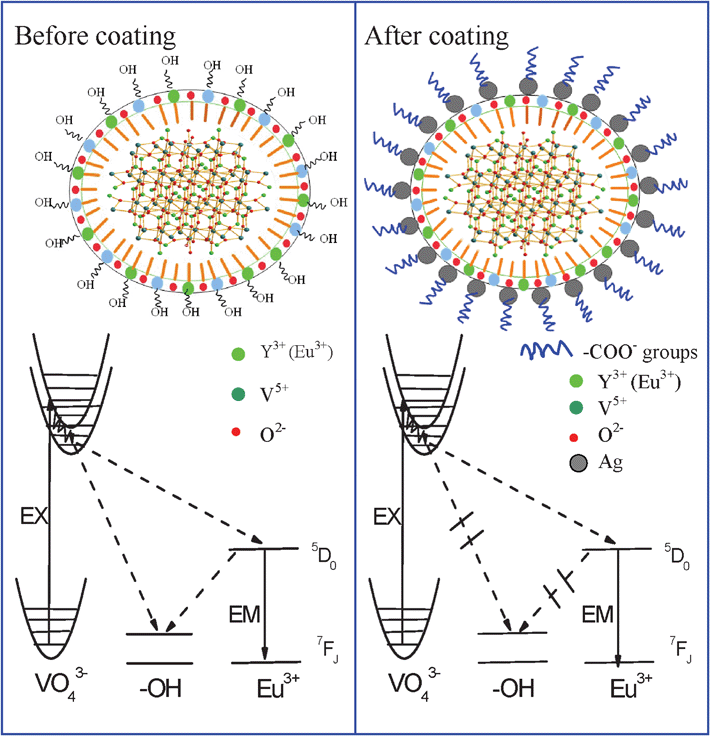 | ||
| Fig. 8 Schematic illustration of ET and PL and luminescent enhancement processes of YVO4:Eu3+ NPs and YVO4:Eu3+@ Ag hybrids. | ||
| Ag +(μM) | Y | Y+Ag+ | 0 | 5 | 16 | 60 | 100 |
| Zeta(mv) | 0.85 | 0.89 | −1.37 | −2.37 | −2.58 | −2.11 | −2.02 |
| En-factor | 1.0 | 1.4 | 2.0 | 9.8 | 8.9 | 7.5 | 5.3 |
Finally, it should be further pointed out that such a highly luminescent enhancement was also observed on the YVO4:Eu3+@Au composites and the upconversion luminescence of YVO4:Yb3+,Er3+ @Ag colloids. And more, most of nanophosphors should have strong and varied interactions with the solvents due to the complex preparation conditions of nanophosphor@noble metal composites, especially, the introducing of different surfactants. These will induce the change of the surface states of NPs and the interface interaction between NPs and solutions inevitably, leading to luminescent enhancement in different degrees. Therefore, we can claim that the present design for luminescent enhancement of YVO4:Eu3+ will be of quite general nature for nanophosphor@noble metal composites in the solution phase.
IV. Conclusions
In this work, a facile seed-mediated growth of YVO4:Eu3+@Ag core-shell hybrid nanostructures colloids was realized. In the YVO4:Eu3+@Ag colloids, the luminescent enhancement of Eu3+ with a highest factor of ∼one order of magnitude was observed when the amount of silver nitrate was 5 μM and the concentration of the YVO4:Eu3+ colloids was 0.07 mg ml−1. The enhancement factor decreased with further increase of the amount of silver NPs, which was caused by the scattering and reflection of silver NPs to excitation light. The luminescent decay dynamics was also systematically studied, and the results demonstrated that the decay lifetime constants of the 5D0–7F2 transitions gradually increased with the increase of silver nitrate and the increase in the YVO4:Eu3+/YVO4:Eu3+@Ag NP concentration. The luminescent mechanism was discussed based on various experiments, which indicated that the main mechanism for the luminescent enhancement in YVO4:Eu3+@Ag hybrids colloids originated from interface effect, rather than the surface plasmon induced absorption enhancement of emitters and/or the increase of radiative decay rate of emitters due to the resonance of the emission with the surface plasmon of silver NPs. In the YVO4:Eu3+@Ag hybrids, the nonradiative ET from the NPs to OH bonds of the solution was hindered considerably. A detailed model was proposed to explain the interesting luminescence enhancement behavior. This work is significant in obtaining efficient RE-doped hybrid colloids and aiding the understanding of the interaction of RE ions with noble NPs.Acknowledgements
The authors would like to acknowledge National Talent Youth Science Foundation of China (Grant No.60925018),and the National Natural Science Foundation of China (Grant Nos. 20971051, 51002062, 11174111, and 61177042).References
- (a) A. J. Kenyon, Prog. Quantum Electron., 2002, 26, 225–284 CrossRef CAS; (b) D. K. Matsuura, Appl. Phys. Lett., 2002, 81, 4526–4528 CrossRef CAS.
- (a) S. Q. Xu, H. P. Ma, D. W. Fang, Z. X. Zhang and Z. H. Jiang, Mater. Lett., 2005, 59, 3066–3068 CrossRef CAS; (b) J. Zhang, S. W. Wang, T. J. Rong and L. D. Chen, J. Am. Ceram. Soc., 2004, 87, 1072–1075 CrossRef CAS.
- (a) M. Nyk, R. Kumar and Y. Tymish, Nano Lett., 2008, 8, 3834–3838 CrossRef CAS; (b) H. Hu, M. G. Yu, F. Y. Li, Z. G. Chen, X. Gao, L. Q. Xiong and C. H. Huang, Chem. Mater., 2008, 20, 7003–7009 CrossRef CAS; (c) Z. G. Chen, H. L. Chen, H. Hu, M. X. Yu and F. Y. Li, J. Am. Chem. Soc., 2008, 130, 3023–3029 CrossRef CAS; (d) C. Louis, R. Bazzi and A. Christophe, Chem. Mater., 2005, 17, 1673–1682 CrossRef CAS; (e) F. Zhang, R. C. Haushalter, R. W. Haushalter, Y. F. Shi, Y. C. Zhang and G. D. Stucky, Small, 2011, 7, 1972–1976 CrossRef CAS.
- K. Kawano, B. C. Hong, K. Sakamoto, T. Tsuboi and H. J. Seo, Opt. Mater., 2009, 31, 1353–1356 CrossRef CAS.
- G. S. Y.i, H. C. Lu, S. Y. Zhao, Y. Ge, W. J. Yang, D. P. Chen and L. H. Guo, Nano Lett., 2004, 4, 2191–2196 CrossRef CAS.
- S. F. Lim, R. Riehn, W. S. Ryu, N. Khanarian, C. K. Tung, D. Tank and R. H. Austin, Nano Lett., 2006, 6, 169–174 CrossRef CAS.
- K. W. Krämer, D. Biner, G. Frei, H. U. Güdel, M. P. Hehlen and S. R. Lüthi, Chem. Mater., 2004, 16, 1244–1251 CrossRef.
- J. Shen, L. D. Sun and C. H. Yan, Dalton Trans., 2008, 5687–5697 RSC.
- (a) D. Q. Chen, Y. L. Yu, Y. S. Wang, P. Huang and F. Y. Weng, J. Phys. Chem. C, 2009, 113, 6406–6410 CrossRef CAS; (b) M. B. Xie, Y. Tao, Y. Huang, H. B. Liang and Q. Su, Inorg. Chem., 2010, 49, 11317–11324 CrossRef CAS.
- (a) H. X. Mai, Y. W. Zhang, R. Si, Z. G. Yan, L. D. Sun, L. P. You and C. H. Yan, J. Am. Chem. Soc., 2006, 128, 6426–6436 CrossRef CAS; (b) J. C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444–7445 CrossRef CAS.
- (a) K. Riwotzki and M. J. Haase, J. Phys. Chem. B, 1998, 102, 10129–10135 CrossRef CAS.
- (a) N. Liu, W. P. Qin, G. S. Qin, T. Jiang and D. Zhao, Chem. Commun., 2011, 47, 7671–7673 RSC; (b) W. Feng, L. D. Sun and C. H. Yan, Chem. Commun., 2009, 4393–4395 RSC.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS.
- (a) T. Hayakawa, S. T. Selvan and M. Nogami, J. Non-Cryst. Solids, 1999, 259, 16–22 CrossRef CAS; (b) L. P. Naranjo, Appl. Phys. Lett., 2005, 7, 241914–241916 CrossRef; (c) T. Hayakawa, S. T. Selvan and M. Nogami, Appl. Phys. Lett., 1999, 74, 1513–151 CrossRef CAS; (d) F. Zhang, G. B. Braun, Y. F. Shi, Y. C. Zhang, X. H. Sun, N. O. Reich, D. Y. Zhao and G. Stucky, J. Am. Chem. Soc., 2010, 132, 2850–2851 CrossRef CAS; (e) X. N. Fang, H. W. Song, L. P. Xie, Q. Liu, H. Zhang and X. Bai, J. Chem. Phys., 2009, 131, 054506–054512 CrossRef; (f) L. Sudheen dra, V. Ortalan, S. Dey, N. D. Browning and I. M. Kennedy, Chem. Mater., 2011, 23, 2987–2993 CrossRef CAS; (g) W. Deng, L. Sudhe en dra, J. B. Zhao1, J. X. Fu, D.Y. Jin, I. M. Kennedy and E. M. Goldys, Nanotechnology, 2011, 22, 325604–3256011 CrossRef.
- C. Louis, S. Roux, G. Ledoux, L. Lemelle, P. Gillet, O. Tillement and P. Perriat, Adv. Mater., 2004, 16, 2163–2166 CrossRef CAS.
- (a) J. Q. Gu, L. D. Sun, Z. G. Yan and C. H. Yan, Chem.–Asian J., 2008, 3, 1857–1864 CrossRef CAS; (b) Z. Q. Li, L. M. Wang, Z. Y. Wang, X. H. Liu and Y. J. Xiong, J. Phys. Chem. C, 2011, 115, 3291–3296 CrossRef CAS; (c) M. Wang†, W. Hou†, C. C. Mi, W. X. Wang, Z. R. Xu, H. H. Teng, C. B. Mao and S. K. Xu, Anal. Chem., 2009, 81, 8783–8789 CrossRef; (d) J. Q. Gu, J. Shen, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2009, 112, 6589–6593 CrossRef.
- H. Zhang, Y. J. Li, I. A. Ivanov, Y. Q. Qu, Y. Huang and X. F. Duan, Angew. Chem. Int. Ed., 2010, 49, 2865–2868 CAS.
- C. Hsu and R. C. Powell, J. Lumin., 1975, 10, 273–293 CrossRef CAS.
- J. H. Zhang, J. B. Liu, S. Z. Wang, P. Zhan, Z. L. Wang and N. B. Ming, Adv. Funct. Mater., 2004, 14, 1089–1094 CrossRef CAS.
- W. X.u, Y. Wang, X. Bai, B. Dong, Q. Liu, J. S. Chen and H. W. Song, J. Phys. Chem. C, 2010, 114, 14018–14024 CAS.
- L. Cheng, K. Yang, Y. G. Li, J. H. Chen, C. Wang, M. W. Shao, S. T. Lee and Z. Liu, Angew. Chem., Int. Ed., 2011, 50, 7385–7390 CrossRef CAS.
- L. P. Xie, H. W. Song, Y. Wang, W. Xu, X. Bai and B. Dong, J. Phys. Chem. C, 2010, 114, 9975–9980 CAS.
- K. Riwotzki and M. Haase, J. Phys. Chem. B, 2001, 105, 12709–12713 CrossRef CAS.
- K. Riwotzki and M. Haase, J. Phys. Chem. B, 1998, 102, 10129–10135 CrossRef CAS.
- W. Drozdowski and A. J. Wojtowicz, Nucl. Instrum. Methods Phys. Res., Sect. A, 2002, 486, 412–416 CrossRef CAS.
- H. S. Peng, H. W. Song, B. J. Chen, J. W. Wang, S. Z. Lu, X. G. Kong and J. H. Zhang, J. Chem. Phys., 2003, 118, 3277–3282 CrossRef CAS.
- V. LeBihan, A. Pillonnet, D. Amans, G. Ledoux, O. R. Marty and C. Dujardin, Phys. Rev. B, 2008, 78, 113405–113408 CrossRef.
- G. Mialon, A. Alexandrou, T. Gacoin and J. P. Boilot, J. Phys. Chem. C, 2009, 113, 18699–18706 CAS.
| This journal is © The Royal Society of Chemistry 2012 |