Water-mediated structural tunability of an alkyl/siloxane hybrid: from amorphous material to lamellar structure or bilamellar superstructure†
S. C.
Nunes
abc,
N. J. O.
Silva
d,
J.
Hümmer
a,
R. A. S.
Ferreira
d,
P.
Almeida
ce,
L. D.
Carlos
d and
V.
de Zea Bermudez
*ab
aChemistry Department, University of Trás-os-Montes e Alto Douro, 5001-801, Vila Real, Portugal. E-mail: vbermude@utad.pt; Fax: +351 259 350 480; Tel: +351 275 259 350 253
bCQ-VR, University of Trás-os-Montes e Alto Douro, 5001-801, Vila Real, Portugal. E-mail: vbermude@utad.pt; Fax: +351 259 350 480; Tel: +351 275 259 350 253
cChemistry Department, Faculty of Sciences, University of Beira Interior, 6200-001, Covilhã, Portugal. E-mail: pjsa@ubi.pt; Fax: +351 275 319 730; Tel: +351 275 329 174
dPhysics Department and CICECO, University of Aveiro, 3810-193, Aveiro, Portugal. E-mail: lcarlos@ua.pt; Fax: +351 234 378 197; Tel: +351 234 370 946
eCICS-UBI – Health Sciences Research Center, University of Beira Interior, 6200-001, Covilhã, Portugal. E-mail: pjsa@ubi.pt; Fax: +351 275 319 730; Tel: +351 275 329 174
First published on 16th January 2012
Abstract
Mono-amide cross-linked alkyl/siloxane hybrids (classed as mono-amidosils and represented by the notation m-A(x), where m and A stand for mono and amide, respectively, and x is the number of methylene repeat units) with water-mediated tunable structures have been prepared by means of sol–gel chemistry and self-direct assembly routes from the organosilanes CH3(CH2)x–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)NH–(CH2)3–Si–(OCH2CH3)3. The amorphous sample m-A(8) has been produced under the stoichiometric conditions (molar ratio Si
O)NH–(CH2)3–Si–(OCH2CH3)3. The amorphous sample m-A(8) has been produced under the stoichiometric conditions (molar ratio Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol:water = 1:4:2) used previously to obtain the lamellar bilayer highly organized m-A(14) hybrid material, showing that the length of the pendant alkyl chains affects the degree of order of the materials. Three structurally ordered samples identified as AC-m-A(8) (1:y) (where AC represents acid catalysis and y is the number of moles of water per mole of Si) have been obtained using the acid catalyzed hydrolytic route and different water contents (y = 600, 300 and 100). Water plays a unique role in the organized mono-amidosils: it not only reverses the natural tendency of the precursor molecule with x = 8 to yield a disordered material, but it also allows the induction of order, leading to the formation of a lamellar structure exclusively in AC-m-A(8) (1:100). The presence of a higher water content promotes extra ordering in AC-m-A(8) (1:300) and AC-m-A(8) (1:600), where the lamellar structure coexists with a bilamellar superstructure. Upon heating the AC-m-A(8) (1:600) sample to 120 °C and then cooling it to room temperature, the lamellar structure remained unaffected, while the superstructure was destroyed. The occurrence of the superstructure, although basically associated with the preferential entrapment of water molecules every two lamellae at the siliceous nanodomains, is also correlated with the amide–amide hydrogen bonded array. The m-A(8) and AC-m-A(8) (1:600) hybrids are multi-wavelength emitters under UV/VIS excitation. The emission spectra exhibit a broad band (300–650 nm) resulting from two contributions: a “blue” band, due to electron–hole recombinations in the NH/CO groups of the amide cross-links, and a “purplish-blue” band, due to oxygen-related defects •O–O–Si≡(CO2) in the siliceous nanoclusters. The absolute emission quantum yields determined for m-A(8) and AC-m-A(8) (1:600) in the 360–380 nm excitation wavelength range were 0.10 ± 0.01 and 0.15 ± 0.01, respectively. While the lifetime values of the high and low wavelength components of the siliceous-related emission are practically the same for both samples (∼3.5 ms), the lifetime value of the NH/CO component for m-A(8) is higher than that of AC-m-A(8) (1:600) (∼292 and ∼156 ms, respectively), suggesting a lower non-radiative transition probability for the former hybrid.
:ethanol:water = 1:4:2) used previously to obtain the lamellar bilayer highly organized m-A(14) hybrid material, showing that the length of the pendant alkyl chains affects the degree of order of the materials. Three structurally ordered samples identified as AC-m-A(8) (1:y) (where AC represents acid catalysis and y is the number of moles of water per mole of Si) have been obtained using the acid catalyzed hydrolytic route and different water contents (y = 600, 300 and 100). Water plays a unique role in the organized mono-amidosils: it not only reverses the natural tendency of the precursor molecule with x = 8 to yield a disordered material, but it also allows the induction of order, leading to the formation of a lamellar structure exclusively in AC-m-A(8) (1:100). The presence of a higher water content promotes extra ordering in AC-m-A(8) (1:300) and AC-m-A(8) (1:600), where the lamellar structure coexists with a bilamellar superstructure. Upon heating the AC-m-A(8) (1:600) sample to 120 °C and then cooling it to room temperature, the lamellar structure remained unaffected, while the superstructure was destroyed. The occurrence of the superstructure, although basically associated with the preferential entrapment of water molecules every two lamellae at the siliceous nanodomains, is also correlated with the amide–amide hydrogen bonded array. The m-A(8) and AC-m-A(8) (1:600) hybrids are multi-wavelength emitters under UV/VIS excitation. The emission spectra exhibit a broad band (300–650 nm) resulting from two contributions: a “blue” band, due to electron–hole recombinations in the NH/CO groups of the amide cross-links, and a “purplish-blue” band, due to oxygen-related defects •O–O–Si≡(CO2) in the siliceous nanoclusters. The absolute emission quantum yields determined for m-A(8) and AC-m-A(8) (1:600) in the 360–380 nm excitation wavelength range were 0.10 ± 0.01 and 0.15 ± 0.01, respectively. While the lifetime values of the high and low wavelength components of the siliceous-related emission are practically the same for both samples (∼3.5 ms), the lifetime value of the NH/CO component for m-A(8) is higher than that of AC-m-A(8) (1:600) (∼292 and ∼156 ms, respectively), suggesting a lower non-radiative transition probability for the former hybrid.
Introduction
Advanced multifunctional silica-based organic–inorganic hybrid systems1 prepared through the versatile sol–gel technique2 have attracted considerable interest because of the wide range of potential technological applications.3The experimental conditions (e.g., water and alcohol contents, nature of the solvent, type of catalyst, pH and temperature) of the sol–gel process primarily dictate the morphology, nanostructure and degree of organization of the hybrids. Typically the hybrids synthesized via classical sol–gel reactions are non-organized, polydisperse in size, locally heterogeneous in terms of chemical composition and exhibit a myriad of macrostructures. Recently the interest of the community studying organic/inorganic hybrids shifted to the creation of organized systems4 exhibiting multi-scale order,5 complex phenomena6 and emergent properties.7 In the last few years the combination of sol–gel routes with self-assembly processes8 in the presence of structure directing agents (or templates) has been extensively explored to produce ordered hybrid networks.9
In 2001, the concept of self-directed assembly in sol–gel derived bridged silsesquioxane hybrid materials was first introduced.10 This challenging new approach relies on the fact that in bridged organosilanes M3–Si–(CH2)3–Y–R′–Y–(CH2)3–Si–M3 (where M = OR, Y = urea (–NHC(O)NH–) group, R = alkyl group and R′ = organic group) the growing supramolecular architecture itself plays the role of internal template, directing the organization of the hybrid silica under controlled hydrolysis.11 In these systems the self-assembly driving forces are weak interactions (hydrogen bonds, π–π and/or hydrophobic interactions) established between the Y groups and between the R′ groups. Hierarchically structured ordered architectures with well defined morphologies at the macroscopic scale result. The self-directed assembly of lamellar packed structures was extensively investigated.10,11b,11c,12,13 Long-range ordered lamellar di-urea cross-linked alkyl-bridged silsesquioxanes with variable interlamellar distances were developed through a self-assembly process based on hydrogen bonding between urea groups and hydrophobic interactions between alkyl chains of variable length.10,11b,11c In these systems self-directed assembly is deeply influenced by the synthetic procedure: acid-catalyzed hydrolysis induced the formation of crystalline lamellar structures, whereas fluoride (F−)-catalyzed hydrolysis under stoichiometric conditions yielded entirely amorphous materials. Hybrids with helical morphology were also created from a chiral silylated diureidocyclohexyl derivative via self-directed assembly.14 Chirality transcription from the pure enantiomer precursors depended on the experimental conditions. While an acidic medium yielded right- or left-handed helical fibres from the pure enantiomers or a featureless granular solid from the racemic mixture of the precursors, F−-based catalysis yielded hollow tubular and spherical silica from the enantiomerically pure compounds and the racemic mixture, respectively.14 Hexagonal bidimensional structures15 and ladder-type superstructures16 were also produced by self-directed assembly.
Reports of self-directed assembly in organosilanes M′3–Si–R′′ (where M′ = M or Cl and R′′ = (CH2)nCH3) are scarce.17–20 In these sort of systems the self-directed assembly strategy is based on the fact that the organophilic precursors become amphiphilic as the alkoxyl or chloride groups are replaced by hydrophilic silanol groups upon hydrolysis. Alkylsilanes adopt a bimolecular layered pattern of stacks of alkyl chains perpendicular to the layers. The poly(octadecylsiloxane)s prepared from n-alkyltrichlorosilanes18 and the hybrids synthesised from triethoxy-19 and trimethoxy13a-based alkylsilanes are the only published examples of bilayered lamellar packing in alkylsilanes. Hydrophobic interactions between the organic chains are the main self-assembly force driving the formation of these van der Waals packing structures.
In 2007, our group introduced a hierarchically-structured bilayer amide cross-linked alkyl/siloxane room-temperature white-light emitter hybrid from the organosilane M′3–Si–(CH2)3–Y′–R′′, where M′ = OCH2CH3, Y′ = amide (–NHC(O)–) group and R′′ = (CH2)xCH3 with x = 14 (Scheme S1, ESI†).21 This hybrid, a member of a new class of materials named mono-amidosils and represented by the notation m-A(x) (where m and A stand for mono and amide, respectively, and x is the number of methylene repeat units), is the first example of a photoluminescent bilayered suprastructure displaying unique nanoscopic sensitivity. In this material, self-directed assembly is governed by intermolecular hydrogen bonding between amide groups, tail-to-tail van der Waals interactions between all-transmethyl-terminated alkyl chains assuming a partially interdigitated packing mode, and an entropic term related to the phase separation between the alkyl chains and the siloxane nanodomains.
In m-A(14) the self-assembly forces determine the emergence of a thermally-actuated photoluminescence (PL) memory effect induced by a reversible order–disorder phase transition of the alkyl chains via a series of heating/cooling cycles operating between 23–120 °C. The onset of this transition occurs at ∼90 °C, involving a coherence length L of 150 nm, which corresponds to ∼30 times the distance between the nanodomains (l = 5 nm). The hydrogen-bonded amide–amide network, which comprises ∼33 adjacent cross-linkages with a coherence length L ∼ 15 nm, begins to be disrupted at ∼67 °C,21i.e., before the onset of the order–disorder phase transition. Cooling the system down to room temperature reveals that a strongly temperature-dependent kinetics of reformation much slower (from 10 to 100 h) than the characteristic time scale at which the phase transition occurs (a few minutes).21c The PL is responsive to the annihilation/formation of the hydrogen-bonded amide–amide array exhibiting a unique nanoscopic sensitivity, in contrast with the customary case for which the local environment around the emitter probe determines the emission features. The formation/annihilation of the hydrogen-bonded amide–amide network therefore plays a central role in the emergence of complexity in m-A(14).21a,21b
With the primary goal of creating another mono-amidosil bilayer suprastructure exhibiting a lower order–disorder phase transition temperature than m-A(14) and hence a greater ability to withstand successive heating/cooling cycles with little or no loss of properties, in the present work the shorter chain mono-amidosil m-A(8) has been prepared. In an attempt to reverse the natural tendency of the non-hydrolized mono-amidepropyltriethoxysilane (m-ADPTES(8)) precursor molecule (see Experimental section and Scheme S1, ESI†) to yield a disordered material and in order to produce it as an organized material structurally resembling m-A(14), the synthetic strategy proposed earlier for lamellar bridged silsesquioxanes has been adopted, which involves the addition of a large excess of water (molar ratio Si:H2O = 1:300) instead of the stoichiometric amount and also the use of hydrochloric acid (HCl) as a catalyst.10,11b,11c In the present case samples with molar ratios Si:H2O = 1:600, 1:300 and 1:100 have been synthesized. For comparison purposes, the m-A(14) mono-amidosil has also been prepared using the same stoichiometric reaction conditions employed previously (molar ratio Si:H2O:ethanol = 1:2:4).21a,21b The ultimate objective of this work has been to investigate in depth the structure, morphology and phototoluminescence (PL) features of the mono-amidosil samples synthesized. Several characteristics of m-A(14) not reported previously have also been examined in the framework of this study. Finally we would like to note that the study of the properties of the m-A(8)-type samples as a function of temperature falls outside the scope of the present work.
Experimental section
Synthesis of m-ADPTES(x) mono-amidosil precursors with x = 8 and 14
To synthesize m-ADPTES(8) mono-amidosil a volume of decanoyl chloride (Cl–C(O)–(CH2)8CH3, DC, Aldrich) was added to a solution prepared through the addition of 3-aminopropyltriethoxysilane (CH3CH2O)3Si(CH2)3NH2, APTES, Fluka)) to a tetrahydrofuran (THF, Riedel de Haën, water free) solution containing pyridine (py, Sigma-Aldrich) and Amberlyst A-21 Ion-Exchange Resin (4.7 meq g−1) (molar ratio APTES:DC:py:resin = 1:1:0.2:1.2) (Scheme S1, ESI†). Additional details of the synthesis of the two m-ADPTES(x) mono-amidosil precursors have been collected in Table S1 of the ESI†. The resulting mixture was sealed and stirred under nitrogen for 5 h at room temperature. The intensity of the FT-IR band attributed to the stretching vibration of the CO group of DC, at 1800 cm−1 was progressively reduced, until it disappeared upon completion of the reaction. In parallel, a series of new bands, associated with the vibrations of the amide group, appeared in the 1760–1530 cm−1 region. The solution was filtrated and the solvent evaporated. The m-ADPTES(8) mono-amidosil precursor, obtained as a transparent oil, was dried under vacuum for several hours. Its structure was confirmed by means of 1H NMR (400.13 MHz, CDCl3) and 13C NMR (100.62 MHz, CDCl3),19,22 and FT-IR (Table S2, ESI†). The m-ADPTES(14) material was obtained according to the procedure reported previously.21a,21b
Synthesis of mono-amidosils via the classical route (m-A(x) with x = 8 and 14)
The synthesis of the m-A(8) mono-amidosil was accomplished using the method described in detail elsewhere for m-A(14).21a,21b A mixture of ethanol (CH3CH2OH, Merck) and bidistilled water (molar ratio APTES:CH3CH2OH:H2O = 1:4:2) was added to m-ADPTES(8) to promote the hydrolysis and condensation reactions characteristic of the sol–gel process (Table S1 and Scheme S1, ESI†). Unlike the m-A(14) mono-amidosil hybrid, which was produced as a white powder,21 the m-A(8) mono-amidosil hybrid was obtained as a transparent, rigid monolith with a yellowish hue.
Synthesis of mono-amidosils via the acid-catalized hydrolytic route (AC-m-A(8) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :y))
:y))
An aqueous solution of hydrochloric acid (1 M, HCl, Riedel de Haën) was added to the m-ADPTES(8) precursor (molar ratios m-ADPTES(8):H2O (or y):HCl = 1:600:0.2; 1:300:0.2 and 1:100:0.1). The resulting mixture was stirred in an oil-bath (∼70–80 °C) for one week. The white slurry solution produced was filtered. The three white solids obtained (AC-m-A(8) (1:600), AC-m-A(8) (1:300) and AC-m-A(8) (1:100)) were washed with a large excess of bidistilled water, ethanol and acetone, and subsequently aged for a period of two weeks in an oven at about 50 °C. Details of the synthetic procedure can be found in Table S1 of the ESI†.
29Si Magic Angle Spinning (MAS) and 13C Cross Polarization (CP)/MAS NMR spectra were recorded on a Bruker Avance 400 (9.4 T) spectrometer at 79.49 and 100.62 MHz, respectively. 29Si MAS NMR spectra were recorded with 2 μs (θ ∼ 30°) rf pulses, and recycle delay of 60 s and at a 5.0 kHz spinning rate. 13C CP/MAS NMR spectra were recorded with 4 μs 1H 90° pulse, 2 ms contact time, a recycle delay of 4 s and at a spinning rate of 8 kHz. The δ are quoted in ppm from TMS.
The X-ray diffraction (XRD) patterns were recorded with a Philips X'Pert MPD powder X-ray diffractometer, using monochromated Cu-Kα radiation (λ = 1.54 Å) over a q range between 0.77 and 25 nm−1 (q = 4πsinθ/λ, 2θ being the scattering angle). The low q region was also recorded at the National Synchrotron Light Laboratory (LNLS), Campinas (Brazil), using a Small Angle X-ray Scattering (SAXS) beam line which provides a monochromatic (λ = 1.608 Å) and horizontally focused beam. The parasitic scattering intensity from air, slits, and windows was subtracted from the total intensity. The scattering intensity was also normalized by taking into account the varying intensity of the direct X-ray beam, sample absorption and sample thickness. The integral breadth of the first order diffraction peak (B) observed in the Small Angle X-ray Scattering (SAXS) pattern was corrected from the instrumental broadening (b) by using highly crystalline poly(oxyethylene)23 as a standard reference of experimental aberrations. The corrected integral breadth was estimated from β2 = B2 − b2.24
High Resolution Scanning Electronic Microscopy (HR-SEM) micrographs were obtained using a FEI Quanta 400FEG/Edax Genesis X4M scanning electron microscope. Elemental analysis on microscopic sections of the samples were performed by Energy Dispersive Spectroscopy (EDS). SEM images were obtained at high or low vacuum, depending on sample charging effects, and in the secondary electrons mode or backscattered electrons mode.
Transmission Electronic Microscopy (TEM) micrographs were obtained using a LEO 906 E Leica microscope. The TEM images were printed as photographs using two types of KODAK Electron film negatives (4489.8 cm × 10 cm or 6.5 cm × 9 cm).
Atomic Force Microscopy (AFM) images were recorded in a Veeco Metrology Multimode/Nanoscope IVA equipment (CEMUP-Porto contract REEQ/1062/CTM/2005), in tapping mode using a super sharp silicon tip, curvature radius 10 nm, and frequency resonance equals to ∼300 kHz. Flattening and elimination of line noise tools and a Lowpass filter provided by the WSXM software25 were used to improve the quality of the images.
Polarized Optical Microscopy (POM) images were recorded using an OPTIKA B-600POL microscope equipped with a 8 Mpixel digital Photo Camera. The images were analyzed using the OPTIKA Vision Pro software.
The thermal behaviour of the m-A(8) mono-amidosil was studied by means of Differential Scanning Calorimetry (DSC) using with a DSC131 Setaram equipment. A mass of approximately 10–20 mg of the sample was placed in a 30 μl aluminium can and stored in a dessicator over phosphorous pentoxide for one week at room temperature under vacuum. After the drying treatment the can was hermetically sealed and the thermogram was recorded. The sample was heated from 25 to 250 °C at 10 °C min−1. The purge gas used in the experiment was high purity nitrogen (N2) (35 cm3 min−1). Time-dependent DSC measurements of the AC-m-A(8) material were carried out using a DSC-50 Shimadzu equipment. A mass of approximately 6.3 mg of the sample was heated at 10 °C min−1 from 25 up to 130 °C, cooled back to room temperature and stored at this temperature for a month. After this period of time it was heated again to 120 °C at the same heating rate indicated above. The purge gas used was high purity N2 (20 cm3 min−1).
Samples for thermogravimetric analysis (TGA) were transferred to open platinum crucibles and analysed using a Q50 TA thermogravimetric analyzer at a heating rate of 10 °C min−1 using dried N2 as purging gas (40 and 60 cm3 min−1 in the balance and in the sample, respectively).
Fourier Transform infrared (FT-IR) spectra were obtained with solid samples (2 mg) which were finely ground, mixed with approximately 175 mg of dried potassium bromide (KBr, Merck, spectroscopic grade) and pressed into pellets. Prior to recording the spectra, the pellets were first vacuum dried at 80–95 °C for about 35–60 h, in order to reduce the levels of adsorbed water and solvent, and then transferred into a glove-box. The FT-IR spectra were acquired a room temperature using a Bruker 22 (Vektor) spectrometer placed inside a glove-box with a dry argon atmosphere. The spectra were collected over the 4000–400 cm−1 range by averaging 150–200 scans at a spectral resolution of 2 cm−1.
The Fourier Transform Raman (FT-Raman) spectra were recorded at room temperature with a Bruker IFS-66 spectrometer equipped with a FRA-106 Raman module and a near-infrared YAG laser with wavelength 1064 nm. The spectra were collected over the 3200–300 cm−1 range at a resolution of 2 cm−1. The accumulation time for each spectrum was 4 h.
To evaluate complex FT-IR and FT-Raman band envelopes and to identify underlying spectral components, the iterative least-squares curve-fitting procedure in the PeakFit software (version 4)26 was used extensively throughout this study. The best fit of the experimental data was obtained by varying the frequency, bandwidth and intensity of the bands. Because of the morphology of the materials under investigation, we employed Voight band functions in the case of the m-A(8) sample and Lorentzian band functions in the case of the m-A(14) and AC-m-A(8) (1:600) mono-amidosils.
The PL measurements were recorded at 12 K and room temperature, using a Fluorolog-3 (FL3–2T model) with double excitation spectrometer, fitted with a 1200 grooves mm−1 grating blazed at 330 nm, and a single emission spectrometer (TRIAX 320), fitted with a 1200 grooves mm−1 grating blazed at 500 nm, coupled to a R928 photomultiplier. The excitation source was a 450 W Xe arc lamp. Excitation spectra were corrected from 240 to 600 nm for the spectral distribution of the lamp intensity using a photodiode reference detector. Emission spectra were also corrected for the spectral response of the monochromators and the detector using typical correction spectra provided by the manufacturer. The quantum yields were measured at room temperature using a quantum yield measurement system C9920-02 from Hamamatsu with a 150 W Xenon lamp coupled to a monochromator for wavelength discrimination, an integrating sphere as sample chamber and a multi channel analyzer for signal detection. Three measurements were made for each sample so that the average value is reported. The method is accurate within 10%.
Results and discussion
The 13C CP/MAS NMR spectra (Fig. S1(A), ESI†) and the 29Si MAS NMR spectra (Fig. S1(B), ESI†) of the m-A(8) and AC-m-A(8) (1:600) mono-amidosils were used to characterize their chemical structure and to determine the degree of conformational disorder of the pendant alkyl chains. The δ and assignment19,21,22c,27 of the resonance peaks exhibited by these NMR spectra are given in Table S3 of the ESI†. For comparison purposes, the 13C CP/MAS and 29Si MAS NMR spectra of m-A(14) have been also included in Fig. S1(A) and S1(B) of the ESI†, respectively, and the most relevant NMR data have been introduced in Table S3 of the ESI†.
The 13C CP/MAS spectrum of the m-A(8) mono-amidosil is dominated by a peak at 30 ppm (Fig. S1(A)(a), ESI†), an indication of the existence of a major proportion of disordered alkyl chains (gauche conformations).18,28 In contrast, the most prominent peak of the 13C CP/MAS spectrum of the AC-m-A(8) (1:600) hybrid is located at 33 ppm (Fig. S1(A)(b), ESI†), a situation similar to that encountered in the case of m-A(14)21 (Fig.S1(A)(c), ESI†). This finding reveals that the great majority of the alkyl chains of AC-m-A(8) (1:600) adopt all-trans zigzag conformations and are thus highly ordered.18,19,29 Comparison of the resonances of m-A(8) and AC-m-A(8) (1:600) (Table S3, ESI†) with those of the m-ADPTES(8) precursor (Table S2, ESI†) confirms that no cleavage of the functional groups of the latter molecule (propyl and alkyl chains and amide cross-links) occurred during the second stage of the synthetic procedure. Moreover, the absence of the peaks associated with the resonance of the ethoxyl groups in the 13C CP/MAS spectra of m-A(8) and AC-m-A(8) (1:600) allows us to infer that the hydrolysis reaction was complete in both hybrid samples (Table S3, ESI†).
The 29Si MAS NMR spectra of m-A(8) and AC-m-A(8) (1:600) (Fig. S1(B), ESI†) exhibit three signals at approximately −52, −59 and −67 ppm, assigned to the T1 (CH2–Si(OSi)(OR)2), T2 (CH2–Si(OSi)2(OR)) and T3 (CH2–Si(OSi)3) substructures, respectively (Table S3, ESI†). The absence of T0- and Q-type sites confirms that in both mono-amidosils no precursor was left unreacted and that the Si–C bonds were preserved, respectively. Although in both materials the main silicon environments present are T2 and T3, in AC-m-A(8) (1:600) the relative proportion of T2/T3 sites is considerably higher than in m-A(8). Table S3 in the ESI† shows that, while the polycondensation degree c calculated for m-A(8) is 83%, the value found for AC-m-A(8) (1:600) is 73%, thus, coincident with that deduced previously for m-A(14).21a,21b The magnitude of c in the latter material is indicative of the presence of 2D siliceous networks. The empirical formulae derived for m-A(8) and AC-m-A(8) (1:600) from the 29Si MAS NMR data reveals that some hydroxyl groups persist bonded to the silicon atoms (Table S3, ESI†).
The XRD patterns reproduced in Fig. 1A demonstrate that the acid-catalyzed hydrolytic route leads to a considerably more ordered material (AC-m-A(8) (1:600)) than non-catalyzed stoichiometric conditions (m-A(8)). Indeed, the pattern of m-A(8) (Fig. 1A(a)) is typical of amorphous materials, with a broad peak centred at ∼14.5 nm−1 (including contributions associated with ordering within the 2D siliceous domains,30 chain–chain31 and amide–amide spacings11b) and a second peak in the low-q range at 2.36 nm−1, assigned to scattering interference between the siliceous regions separated by the alkyl chains30,32 with an average inter-region distance of 2.66 nm. In contrast, the XRD pattern of AC-m-A(8) (1:600) (Fig. 1A(b)) exhibits a narrower peak in the high-q region centred near 14.75 nm−1, in which the above mentioned contributions are more deconvoluted, and a series of reflections in the low-q region which reveal the presence of medium- to long-range ordered structures. The most intense peak detected at about q = 1.94 nm−1 in the XRD and SAXS patterns (black and red curves of Fig. 1B, respectively) represents the fundamental order of a lamellar bilayer structure with an interlamellar spacing l1 = 3.24 nm (where l = k2π/qk), a value lower than that reported for m-A(14) (l = 5.0 nm).21a,21b This was an expected result because the alkyl chains of AC-m-A(8) (1:600) are shorter than those of m-A(14). The peaks associated with this ordering have been marked with continuous vertical lines in Fig. 1B. An estimation of the interlamellar spacing l1 can be made considering the average distances of the bonds present in each pendant alkyl chain.21 Assuming average distances of 0.13 nm for the –C–C–, –C–N–, –N–C(O)– and –C(O)–C– bonds and considering that each pendant alkyl chain contains 3 propyl carbon atoms, 1 amide nitrogen atom, 1 carbonyl carbon atom and 9 alkyl carbon atoms, a theoretical chain length of 1.69 nm and a maximum interlamellar distance of 3.38 nm result. The latter value, very close to the experimental one (l1 = 3.24 nm), demonstrates that the degree of interdigitation in AC-m-A(8) (1:600) is very low since only two carbon atoms (i.e., one C–C bond) of opposed chains appear to be interpenetrated, unlike the case of m-A(14) where a much larger interpenetration effect was evident.21a,21b
 | ||
| Fig. 1 A: XRD patterns of the m-A(8) (a), AC-m-A(8) (1:600) (b) and m-A(14)21 (c) mono-amidosils. B: SAXS (red line) and XRD (black line) patterns of AC-m-A(8) (1:600). The blue line represents the XRD pattern obtained after heating AC-m-A(8) (1:600) to 120 °C and cooling it to room temperature. The vertical continuous and doted lines represent the peaks associated with the lamellar structure and the bilamellar superstructure, respectively. C: XRD patterns of AC-m-A(8) (1:100) (a), AC-m-A(8) (1:300) (b) and AC-m-A(8) (1:600) (c). Inset shows zoom over the low-q region. | ||
The HR-SEM image reproduced in Fig. 2(a) shows that the AC-m-A(8) (1:600) mono-amidosil consists of irregular and dense objects. The intrinsic 2D ordered nanostructure of the film, with a characteristic repeating distance of about 4 nm, is evident in the TEM image of Fig. 2(b), corresponding to the interlamellar distance l1. The value found is slightly larger than that obtained from XRD. This may be due to the fact that the TEM distance is not representative of the entire sample. It is likely that in TEM the most structured regions are more easily observed than the less structured ones. Moreover, in the mono-amidosil systems better ordering is associated with larger interlamellar distances (as concluded by comparing the AC-m-A(8) and m-A(8) materials). Evidence of the organized lamellar texture of this sample has been obtained by AFM upon scanning its surface in tapping mode. The ordered organization of AC-m-A(8) (1:600) may be observed in the 3D topographic perspective given in Fig. 2(c). Fig. 3 reveals that the lamellae of m-A(14) adopt a more regular organization. The birefringence of AC-m-A(8) (1:600) detected in the POM image recorded between crossed polarizers (Fig. 2(d)) suggests submicrometric anisotropy.
 | ||
| Fig. 2
HR-SEM (a), TEM (b), 3D-AFM phase (c) and POM (crossed polarizers) (d) images of the AC-m-A(8) (1:600) mono-amidosil. | ||
 | ||
| Fig. 3 3D-AFM image of the m-A(14) mono-amidosil. | ||
Interestingly, the SAXS pattern of AC-m-A(8) (1:600) (red curve of Fig. 1B) shows a peak at about q = 0.95 nm−1, which corresponds to a characteristic distance l2 = 6.41 nm, thus approximately twice as large as l1. This peak is ascribed to the presence of a superlamellar ordering involving two successive lamellar bilayers as the repeating motive, therefore implying an extra ordering in the AC-m-A(8) (1:600) system, when compared to previously reported ordered hybrid materials.11b,21,18 This ordering gives rise to a series of higher order peaks (marked with dotted vertical lines in Fig. 1B) following the relation l = k2π/qk, where even k values produce peaks at the same position of those characteristic of the lamellar order and odd k values generate the peaks associated exclusively with the bilamellar order.
To further characterize the structure of AC-m-A(8) (1:600) and specifically to investigate the origin of the superlamellar arrangement, DSC and ATG measurements were performed.
Complementary XRD data (Fig. 1C) were also obtained with two different AC-m-A(8)-type samples synthesized with lower water contents (AC-m-A(8) (1:300) and AC-m-A(8) (1:100), see Experimental section).
DSC is an extremely powerful technique for studying the energetics of the order/disorder phase transitions of the alkyl chains. Nagle and Goldstein33 noted that during the fusion of alkyl chains most of the enthalpy change is associated with a large cohesive van der Waals energy term, whereas the entropy change involves a conformational term, an excluded volume term and a dominating volume expansion term. The absence of melting peaks in the DSC curve of m-A(8) (Fig. 4(a)) corroborates the conclusion drawn from XRD that this hybrid is entirely amorphous. In contrast, the DSC thermogram of AC-m-A(8) (1:600) (Fig. 4(b)) exhibits two endothermic events: one centered at 87 °C (Tonset = 84 °C) and another one, stronger, broader and ill-defined, centred at ∼108 °C. Considering the chemical similarity between m-A(14) and AC-m-A(8) (1:600) and taking into account that the latter material comprises shorter chains, one would have normally expected to find a single endothermic peak shifted to temperatures lower than 90 °C, which is, as stated already, the onset temperature of the order/disorder phase transition of m-A(14) (Fig. 4(d)).21a,21b We will therefore partly ascribe the low-temperature endotherm of AC-m-A(8) (1:600) to the order/disorder phase transition of the alkyl chains. In an attempt to determine the nature of the high-temperature peak the AC-m-A(8) (1:600) sample was heated to 120 °C (i.e., above the order/disorder phase transition temperature) and then allowed it to cool to room temperature. The DSC curve recorded after this thermal treatment is shown in Fig. 4(c). The disappearance of the high-temperature endothermic peak and the presence of a single endotherm centred at 66 °C may be interpreted as proof of the destruction of the bilamellar superstructure and of the simultaneous preservation of the lamellar structure. The downshift of the low-temperature peak from 86 °C (Fig. 4(b)) to 66 °C (Fig. 4(c)) leads us to suggest that the former thermal event is associated, not only with the order/disorder phase transition, but also with the release of water molecules.
 | ||
| Fig. 4
DSC curves of the mono-amidosils m-A(8) (scale bar = 100 mW g−1) (a), AC-m-A(8) (1:600) (scale bar = 50 m Wg−1) (b), AC-m-A(8) (1:600) after being heated to 130 °C, cooled to room temperature and stored at room temperature for a month (scale bar = 50 mW g−1) (c) and m-A(14) (scale bar = 100 mW g−1)21 (d). | ||
Comparison of the SEM and AFM images of the as-prepared (Fig. 2(a) and (c), respectively) and thermally treated (Fig. 5(a) and (b), respectively) AC-m-(8) (1:600) allows to recognize marked structural changes. In particular, Fig. 5(a) reveals the occurrence of a void-like, considerably less dense structure in the material which was subject to the thermal treatment, a result that may be interpreted as an indication of the breakdown of the superstructure.
 | ||
| Fig. 5
HR-SEM (a), 3D-AFM Plane (b) and POM/crossed polarizers (c) images of the AC-m-A(8) (1:600) mono-amidosil recorded a month after the sample was heated to 120 °C and cooled to room temperature. | ||
The TGA data demonstrate that the thermal degradation of m-A(8) (Fig. 6(a)) and m-A(14) (Fig. 6(c)) is initiated at approximately 250 °C and occurs in a single step involving a major weight loss. The behaviour of AC-m-A(8) (1:600) is completely different, as it already starts to suffer decomposition gradually from 25 °C (Fig. 6(b)). Three weight loss regions may be discerned (see derivative curve and inset of Fig. 6). Regions I and II are characterized by transitions at 99 and 154 °C, respectively, and weight losses of ∼1.15 and 2.69%, respectively. Region III is characterized by several medium intensity transitions plus a large one at 437 °C and a global weight loss of ca 69.2%. In the three samples, 20% (m-A(14) and ∼27% (m-A(8) and AC-m-A(8) (1:600)) of the mass remains undecomposed at 650 °C. Based on the attribution given by Parikh et al. for a similar thermal behavior found in a layered microcrystalline polymer prepared by a hydrolytic polycondensation of n-alkyltrichlorosilanes in water,18 the first two transitions indicated above are assigned to the loss of physically bound water molecules. The weight loss in Region I is most likely related to the release of water molecules entrapped within the siloxane network, whereas the weight loss in Region II is possibly due to the release of water molecules formed through the condensation of silanol groups (cf.29Si MAS NMR data in Table S3 of the ESI†).18 The weight loss taking place in Region III is attributed to the degradation of the alkyl chains, which ends up with a stable (Si–O–Si)-based residue. We suggest that the transition occurring in Region I, corresponding to the release of entrapped water molecules, is intimately correlated with the high-temperature peak discerned in the DSC curve of AC-m-A(8) (1:600) (Fig. 4(b)). Moreover, we propose that water molecules preferentially entrapped every two lamellae are the main reason for the formation of the bilamellar superstructure.
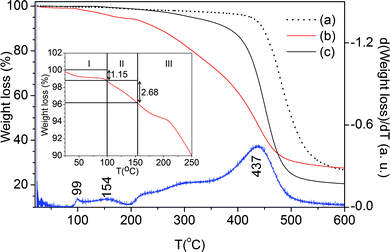 | ||
| Fig. 6
TGA curves of the m-A(8) (a), AC-m-(8) (1:600) (b, red line) and m-A(14)21 (c) mono-amidosils. The blue line corresponds to the derivative curve of (b). The inset shows curve (b) in the 25–200 °C temperature range. | ||
The location of water molecules within the hydrophilic siliceous lamellae is beautifully corroborated by the XRD results, since the interlamellar average distance l1 is larger (i.e., the peaks appear at slightly lower q) in the mono-amidosils with higher water content (Fig. 1C). Moreover, the zoom of the XRD patterns of AC-m-A(8) (1:y) shown in the inset of Fig. 1C reveals that the superlamellar peak (at q ∼ 0.95 nm−1) is present in the samples with higher water content (Fig. 1C(b) and 1C(c)) and practically absent in the sample with the lowest water content (Fig. 1C(a)). This is unequivocal evidence that structural water is surely responsible for the superlamellar arrangement. Here, we suggest that water is selectively present every two lamellae, giving rise to the superlamellar structure (Scheme 1). Further evidence supporting this interpretation comes from the fact that the bilamellar superstructure is destroyed after heating the sample above the temperature where structural water is lost (blue curve in Fig. 1B). This water-mediated ordering process resembles the well known phenomenon (occurring at a lower length scale) of superstructure formation in spinels due to vacancy ordering.34
 | ||
| Scheme 1 Schematic representation of the lamellar and bilamellar structures of the AC-m-(8) (1:600) mono-amidosil. | ||
But the AC-m-A(8) (1:600) material exhibits another very interesting feature. Analysis of the XRD pattern depicted in Fig. 1B reveals that after heating AC-m-A(8) (1:600) to 120 °C and then allowing it to cool to room temperature, the lamellar order is re-established, as in the case of m-A(14),21 whereas the supermolecular ordering is no longer present. At the same time, the high-q region peaks become broader, suggesting that the bilamellar supermolecular ordering is very probably correlated with the 2D siliceous domains, the amide groups and/or chain–chain ordering. These observations provide evidence that both structures (lamellar and bilamellar) have clearly different characteristic recovery times.
These results clearly demonstrate that the synthesis of the m-A(8) mono-amidosil carried out in the absence of a catalyst and using a stoichiometric amount of water (i.e., under the mild classical conditions used previously to obtain the lamellar bilayer highly organized m-A(14) material21) results in an amorphous material. This indicates that a minimum number of methylene groups is required in the pendant alkyl chain (at least more than 8) if one wishes to produce an ordered material using the same experimental conditions. On the other hand, the use of an acid catalyst and the addition of large amounts of water produce an unprecedented improvement of ordering which may even exceed the lamellar structure previously observed in m-A(14) and attain bilamellar superstructuring.
To get further insight into the structure of the as-prepared amorphous m-A(8) and of the lamellar- and bilamellar-structured AC-m-A(8) (1:600), specially in terms of the conformational state of the alkyl chains and of the extent/magnitude of hydrogen bonding, the FT-IR and FT-Raman spectra of both mono-amidosil hybrids were analyzed in depth.
Three vibration modes of the alkyl chains can be employed as probes of their degree of order: (i) The FT-IR and FT-Raman symmetric and asymmetric CH2 stretching modes (νsCH2 and νaCH2, respectively); (ii) The FT-Raman C–C skeletal stretching (νC–C) mode. Details concerning these absorption modes are provided in the ESI†.
The FT-IRνaCH2 and νsCH2 regions of m-A(8) display two intense features at 2929 cm−1 (full width at half maximum (fwhm) = 34 cm−1, intensity = 0.28) and 2856 cm−1 (fwhm = 10 cm−1, intensity = 0.16) (Fig. S2A(a) and Table S4 in the ESI†). The frequency and the fwhm of these bands give support to the explanation that the alkyl chains adopt gauche conformations and are thus essentially amorphous35,36. The FT-IR spectral signature of the AC-m-A(8) (1:600)/m-A(14) mono-hybrids is, however, quite different. The latter samples give rise to two strong bands at 2924 cm−1 (fwhm = 30 cm−1, intensity = 0.95)/2919 cm−1 (fwhm = 22 cm−1, intensity = 2.2) and 2852 cm−1 (fwhm = 15 cm−1, intensity = 0.50) /2850 cm−1 (fwhm = 10 cm−1, intensity = 1.5) (Fig. S2A(b)/Fig. S2A(c) and Table S4 in the ESI†). The frequency, intensity and fwhm of these events indicate that the alkyl chains are fully stretched (all-trans) in the m-A(14) sample, whereas in the case of AC-m-A(8) (1:600), although the population of all-trans conformers is dominant, a significant amount of gauche conformations are also present.35,36
The νCH2 region of the FT-Raman spectra of m-A(8) mono-amidosil exhibits prominent bands at 2929 and 2890 cm−1 and a shoulder at 2951 cm−1 (Fig. S2B(a) and Table S4 in the ESI†). The bands centred at 2951 and 2929 cm−1, characteristic of disordered methylene chains, are ascribed to the νsCH2 fundamental mode and to Fermi resonance between the νsCH2 fundamental with the many overtones of the δCH2 mode.36b,37,38 The intense band at 2890 cm−1 is attributed to the νaCH2 mode of amorphous alkyl chains.39 The FT-Raman spectra of the AC-m-A(8) (1:600) and m-A(14) hybrids (Fig. S2B(b) and Fig. S2B(c), respectively, ESI†) exhibit two intense bands at 2880 and 2845 cm−1 (Table S4, ESI†) that are attributed to the νaCH2 and νsCH2 stretching modes characteristic of the methylene chains in all-trans conformations36b,39 The intensity ratio (r) between the νaCH2 (∼2880 cm−1) and νsCH2 (∼2845 cm−1) bands is sensitive to the conformational disorder of the methylene chains and to their packing arrangement.39 The r value (1.60)21a,21b for m-A(14) mono-amidosil is of the same order of magnitude as those reported for crystalline n-alkanes39 and solid palmitic acid.39 The AC-m-A(8) (1:600) sample leads to a slightly lower r value (1.4). As expected, the value calculated for m-A(8) (1.2) is of the same order of magnitude of the values found for liquids n-alkanes.39 These results support the presence of alkyl chains in only all-trans conformations in the case of m-A(14). In AC-m-A(8) (1:600) a proportion of the alkyl chains is disorganized, meaning that a fraction of gauche conformers coexist with all-trans conformers.
The νC–C region is also sensitive to the relative proportion of gauche and trans conformers present in the alkyl chains. In the FT-Raman in the νC–C region of the m-A(8) mono-amidosil (Fig. S3(a), ESI†) three broad bands are observed at 1121, 1078 and 1064 cm−1. The bands at 1121 and 1064 cm−1 are attributed to the occurrence of trans rotamers.40 The presence of a band at 1080 cm−1 is characteristic of chain randomization (i.e., interruption of all-trans conformations) because of the presence of gauche C–C conformers.37b The FT-Raman spectra of AC-m-A(8) (1:600) and m-A(14) exhibit two prominent bands at approximately 1128 cm−1 and 1062 cm−1 (Fig. S3, ESI†) which are indicative of the occurrence of all-trans conformations.
We may infer from the FT-IR and FT-Raman spectroscopic data discussed above that the alkyl chains of the m-A(8) mono-amidosil are fully disordered and adopt essentially gauche conformations. In contrast, the spectral signature of AC-m-A(8) (1:600) closely resembles that of m-A(14). Indeed, solid evidence was found that in AC-m-A(8) (1:600) the great majority of the alkyl chains adopt all-trans conformations, although a minor proportion of gauche conformers also exists. These conclusions corroborate the 13C CP/MAS NMR and XRD data.
In hybrid samples, such as the mono-amidosils, which incorporate cross-links composed of hydrogen donor groups and hydrogen acceptor groups (NH and CO, respectively) prone to participating actively in hydrogen bonding interactions, the examination of the so-called amide I and amide II bands (see the ESI† for details concerning these absorption modes) is compulsory to determine the extension and strength of hydrogen bonds.
While the FT-IR spectrum of the m-A(8) material is dominated by a sharp amide I band at 1649 cm−1 (Fig. S4(a), ESI†), in the case of AC-m-A(8) (1:600) and m-A(14) this feature emerges at 1640 cm−1 (Fig. S4(b) and S4(c), respectively, ESI†). The intensity maximum of the amide II band is centered at 1522 cm−1 in the FT-IR spectra of the three samples (Fig. S4, ESI†). The frequency difference (Δν) between the intensity maximum of the amide I and amide II bands of the AC-m-A(8) (1:600) (Δν = 88 cm−1) and m-A(14) mono-amidosils (Δν = 87 cm−1)21a,21b is, as expected, lower than that found for the m-A(8) mono-amidosil (Δν = 97 cm−1). These observations indicate, not only that the strength of the hydrogen-bonded arrays in the AC-m-A(8) (1:600) and m-A(14) mono-amidosils is essentially the same, but also that the hydrogen-bonded array of the m-A(8) hybrid is considerably weaker.
Further elucidation of hydrogen bonding interactions in the mono-amidosil samples may be retrieved from the results of curve-fitting performed in the 1725–1500 cm−1 interval (Fig. 7 and Table S4 in the ESI†). The amide I band of m-A(8) was decomposed into four components situated at 1676 (sh, fwhm = 34 cm−1), 1657 (m, fwhm = 24 cm−1), 1643 (s, fwhm = 20 cm−1) and 1626 cm−1 (m, fwhm = 20 cm−1) (Fig. 7(a) and Table S4 in the ESI†). In the case of the m-A(14)/AC-m-A(8) (1:600) hybrids the amide I was decomposed into three components at 1652/1656 (w, fwhm = 27/28 cm−1), 1640/1640 (s, fwhm = 17/20 cm−1) and 1625/1622 cm−1 (w, fwhm = 15/29 cm−1) (Fig. 7(b)/(Fig. 7(c) and Table S4 in the ESI†). The 1676 cm−1 component is attributed to free CO groups21a,21b,41 and the bands at 1657–1652 cm−1 are assigned to the absorption of hydrogen-bonded CO groups in disordered amide–amide aggregates.21a,21b,41,42 Finally, the 1640 and 1626–1622 cm−1 bands are ascribed to the absorption of CO groups included in significantly more ordered hydrogen-bonded amide–amide associations of increasing strength.43 The absence of a band at ∼1676 cm−1 in the amide I band of m-A(14) and AC-m-A(8) (1:600) indicates that no free CO groups exist in both samples41 The amide II band of m-A(8), m-A(14) and AC-m-A(8) (1:600), centred at 1552 cm−1, is less intense than the amide I band (Fig. S4, ESI†). This feature was resolved into two individual components at 1561–1558 cm−1 (fwhm = 24–20 cm−1) and 1546–1542 cm−1 (fwhm = 31–25 cm−1) (Fig. 7 and Table S4 in the ESI†), suggesting that in the three mono-amidosils hydrogen-bonded aggregates with two distinct degrees of order develop. One of the most interesting aspects worth mentioning regards the relative intensity of the amide I components. Fig. 7 and Table S4 in the ESI† reveal that in the AC-m-A(8) (1:600) material the proportion of ordered aggregates is unexpectedly higher than in m-A(14). We are led to conclude from this finding that the bilamellar superordering found in AC-m-A(8) (1:600) is intimately associated with the hydrogen-bonded amide–amide array.
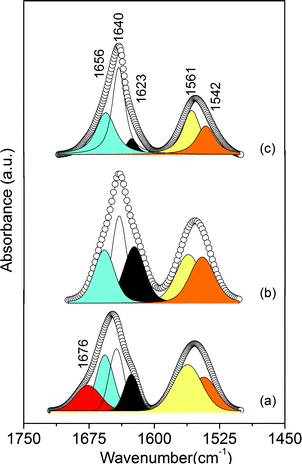 | ||
| Fig. 7 Curve-fitting results in the amide I and amide II regions of the m-A(8) (a), AC-m-(8) (1:600) (b) and m-A(14) (c)21 mono-amidosils. | ||
The m-A(8) and AC-m-A(8) (1:600) mono-amidosils are multi-wavelength emitters under ultraviolet/visible (UV/VIS) excitation in the temperature interval 10–300 K. The room temperature emission spectra reproduced in Fig. 8 show a broad band (300–650 nm), for which the peak position deviates towards the red as the excitation wavelength increases from 330 to 420 nm. Although in this excitation wavelength range the emission and fwhm are independent of the synthetic route, at lower excitation wavelengths (300–330 nm), the emission features of the two samples are distinct. In particular, we note that for AC-m-A(8) (1:600) the emission features are independent of the excitation wavelength being deviated towards the red when compared with those of m-A(8) (Fig. 8). These emission features resemble those previous detected in similar organic–inorganic hybrids, associated with electron–hole recombinations within the NH/CO groups of the cross-linkages and within the siliceous nanoclusters.44,45
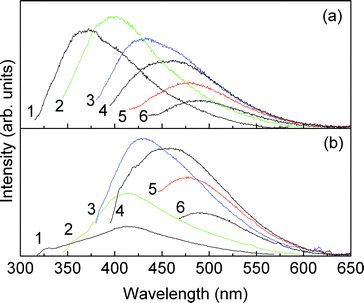 | ||
| Fig. 8 Room temperature emission spectra of m-A(8) (a) and AC-m-A(8) (1:600) (b) excited at (1) 300, (2) 330, (3) 360, (4) 380, (5) 400, and (6) 420 nm. | ||
The absolute emission quantum yield was determined for m-A(8) and AC-m-A(8) (1:600). A maximum and constant value of 0.10 and 0.15 were measured in the excitation range 360–380 nm of these samples, respectively. It is of interest to compare these values with those published in the literature for other amorphous non-doped functionalized hybrids. The quantum yield of m-A(8), although smaller than that of disordered bridged silsesquioxanes (0.24 ± 0.02)46 and di-urethanesils (0.19 ± 0.02),44,45 is higher that the values known for similar di-amide cross-linked alkyl/siloxane hybrids (di-amidosils) (0.05 ± 0.01)22a and for di-urea cross-linked poly(oxyethylene)/siloxane hybrids (di-ureasils) (0.06–0.08 ± 0.01).44 It is noteworthy that for the lamellar mono-amide cross-linked alkyl/siloxane hybrid (mono-amidosil) AC-m-A(8) (1:600) the quantum yield value is similar to that reported for lamellar bridged silsesquioxanes (0.14 ± 0.01),47 but significantly higher than that of the lamellar mono-amidosil m-A(14) (0.03 ± 0.01).21
The PL features of the m-A(8) and AC-m-A(8) (1:600) mono-amidosils were further studied via time-resolved spectroscopy. Fig. 9A and B compare the time-resolved emission spectra acquired for distinct starting delay (SD) values and excitation wavelengths, revealing similar time-resolved emission features for m-A(8) and AC-m-A(8) (1:600). For an excitation wavelength of 290 nm, the emission spectra display a long-live component (Fig. 9A) ascribed to the electron–hole recombinations within the NH/CO groups.44 Increasing the excitation wavelength up to 360 nm, this component deviates towards the red (500 nm) and two components at lower wavelengths, ascribed to the presence of the siliceous domains,44 are clearly discerned. In particular, the emission spectra acquired at SD = 0.05 ms unequivocally reveals the presence of three components at 425, 455 and 498 nm. When the SD value is increased to 5.00 ms only the high-wavelength component remains. Similarly to the situation reported previously for analogous materials, the low-wavelength (425 and 455 nm) and high-wavelength (498 nm) components are due to the presence of electron–hole recombinations within the NH/CO groups of the cross-linkages and within the siliceous nanoclusters.44
 | ||
| Fig. 9
Emission spectra (12 K) of the m-A(8) (a) and AC-m-A(8) (1:600) (b) mono-amidosils, excited at 290 nm (A) and 360 nm (B) acquired at starting delay values of 0.05 ms (black line) and 5.00 ms (red line). The integration window was 20.00 ms. | ||
The higher relative contribution of the siliceous-related components and the unequivocal presence of the component at 455 nm, most particularly evident for m-A(8), is explicitly attributed to the higher condensation degree c and thus lower T2/T3 ratio,44 as pointed out by the 29Si MAS NMR data (Table S2, ESI†).
The lifetime values of the two distinct emission components were selectively monitored under excitation at 360 nm (Fig. S5, ESI†). The emission decay curve associated with the NH/CO groups was monitored at 500 nm with SD = 40.00 ms to avoid the contribution of the faster siliceous-related component as the two emission bands are spectrally overlapped. The siliceous-related emission was monitored at 427 and 455 nm with SD = 0.05 ms. All the emission curves are well fitted by a single exponential function yielding lifetime values for the N–H/CO component of 292.3 ± 9.5 ms and 156.9 ± 12.6 ms for m-A(8) and AC-m-A(8) (1:600), respectively. The high and low wavelength components of the siliceous-related emission display essentially the same lifetime values (3.4 ± 0.2 ms and 3.6 ± 0.3 ms for m-A(8) and AC-m-A(8) (1:600), respectively). The lifetime value of AC-m-A(8) (1:600) is similar to those previously found for the di-ureasils.44 The high lifetime value found for the NH/CO related component in m-A(8) suggests a lower non-radiative transition probability for this hybrid. At room-temperature the time-scale behind the emission features is in the time-range 10−7–10−9 s (not shown), as observed in other analogous organic–inorganic hybrids.44,45
Conclusions
In the present work, we have demonstrated that it is possible to easily control the degree of order of mono-amide cross-linked alkyl/siloxanes (mono-amidosils, m-A(x)) through an adequate choice of the synthetic procedure and/or the length of the pendant alkyl chain.Under the stoichiometric reaction conditions of the classical sol–gel process (i.e., molar ratio Si:H2O:ethanol = 1:2:4), a hierarchically structured lamellar bilayer material is produced when x = 14.21 If x = 8, an amorphous hybrid results instead, an indication that a minimum number of methylene repeat units must exist in the pendant alkyl chain to impart order to the mono-amidosil system. The use of hydrolytic conditions, implying the addition of a large amount of water (and no other solvent) and the incorporation of an acid catalyst (typically HCl), provides a simple way to shift from a naturally occurring amorphous structure (m-A(8) hybrid) to crystalline ones (AC-m-A-(8) (1:y) hybrids).
A possible scenario for the mechanisms that take place in the two mono-amidosils is the following. If the alkyl chains of the precursor are long enough, as it is the case of m-ADPTES(14), the strong intermolecular forces established between the precursor molecules, i.e., hydrogen bonds between the amide groups or between the silanol groups, and van der Waals interactions between the pendant alkyl chains, are solely responsible for the formation and subsequent growth of a supramolecular architecture (composed of lamellar bilayer ribbons) that plays the role of internal template, directing the organization of the hybrid silica. No particular reaction conditions are required and a stoichiometric amount of water may be employed. In the case of m-ADPTES(8), which incorporates chains that are too short, the structuring process needs to be assisted by a large excess of water and the addition of an acid catalyst. These extreme conditions force the pendant alkyl chains to approach, promoting hydrophobic interactions that favor face-to-face stacking of the growing lamellar bilayer ribbons. Because of the low pH of the medium the silanol and amide groups of these growing structured objects become protonated. The high charge mobility and the low charge repulsions that take place in water aid the lamellar stacking.48
Interestingly, based on XRD and SAXS data, we have been able to demonstrate that the degree of order of the AC-m-A-(8) (1:y) samples may be tuned upon incorporation of judicious amounts of water into the reaction medium. While a low water content gives rise to a lamellar structure AC-m-A-(8) (1:100), at high water content (AC-m-A-(8) (1:300) and (AC-m-A-(8) (1:600)), extra ordering is induced and a bilamellar superstructure, with a degree of order that largely exceeds that of the highly organized m-A(14), is formed concomitantly with the lamellar structure. TGA, DSC and XRD/SAXS measurements have allowed us to conclude that this unusual order enhancement effect detected in AC-m-A-(8) (1:300) and AC-m-A-(8) (1:600)) is intimately associated with the entrapment of water molecules every two lamellae of the primary lamellar structure. The water molecules bind to the 2D siliceous network by means of hydrogen bonds established with the silanol groups. The considerable strengthening of the amide–amide hydrogen-bonded array in AC-m-A-(8) (1:600) with respect to m-A(8) detected by FT-IR supports that the creation of the superstructure is a major combined effect in which amide groups are also concerned.
Heating AC-m-A-(8) (1:600) to 120 °C, following by cooling to room temperature, results in the destruction of the superstructure. In contrast, the lamellar structure is preserved. However, reformation of the superstructure takes place after some time (Figure S6, ESI†).
The nanostructured nature of AC-m-A-(8) (1:600) has been confirmed by TEM. Comparison of the SEM images recorded prior and after the thermal treatment to which this sample was subject substantiate the marked changes produced in its morphology. The modifications that might occur in parallel in the topography of the material are less perceptible by AFM.
The amorphous m-A-(8) and the lamellar and bilamellar super-structured AC-m-A-(8) (1:600) mono-amidosils are room temperature white light emitters under UV/VIS excitation. The emission spectra display a broad band composed of a “blue” band, related to electron–hole recombinations in the NH/CO groups of the amide cross-links, and a “purplish-blue” band, due to oxygen-related defects •O–O–Si![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) (CO2) in the siliceous nanodomains. The maximum quantum yields of m-A-(8) and AC-m-A-(8) (1:600) are 0.10 and 0.15, respectively. For the NH/CO component the lifetime values are approximately 292 and 156 ms, respectively. The higher value observed in the case of m-A(8) may be correlated with a lower non-radiative transition probability. With respect to the high and low wavenumber components of the siliceous-related emission both materials exhibit, however, similar lifetime values.
(CO2) in the siliceous nanodomains. The maximum quantum yields of m-A-(8) and AC-m-A-(8) (1:600) are 0.10 and 0.15, respectively. For the NH/CO component the lifetime values are approximately 292 and 156 ms, respectively. The higher value observed in the case of m-A(8) may be correlated with a lower non-radiative transition probability. With respect to the high and low wavenumber components of the siliceous-related emission both materials exhibit, however, similar lifetime values.
This work has clearly opened up extremely interesting prospects for the class of mono-amidosil hybrids. It is clear that several aspects derived from the results reported here need to be investigated in future. The different recovery times exhibited by the lamellar structure and the bilamellar superstructure after submitting the AC-m-A-(8) (1:600) sample to a heating/cooling cycle pointed out by XRD data demand further studies and the use of complementary techniques, such as DSC, TGA, FT-IR, FT-Raman and PL. The influence of super-ordering on the order/disorder phase transition of this material in consecutive heating/cooling cycles deserves a closer look. Such temperature- and time-dependent studies are already underway using FT-IR and PL measurements.
Acknowledgements
This work was supported by Fundação para a Ciência e a Tecnologia (FCT) and FEDER (contracts PTDC/CTM/101324/2008 and PTDC/QUI-QUI/100896/2008). S. C. Nunes acknowledges FCT for a grant (SFRH/BPD/63152/2009). The authors are grateful to Denis Ostrovskii, of the Department of Applied Physics, Chalmers University of Technology, Göteborg (Sweden), for recording the FT-IR and FT-Raman spectra, and to Sidney J. L. Ribeiro, of the Chemistry Institute of Araraquara, UNESP (Brazil), for recording the SAXS patterns at the LNLS, Campinas (Brazil). J. Hümmer, a student of Technology of Functional Materials at the Julius-Maximilian-University in Würzburg (Germany), was involved in this work in the framework of an internship at the Department of Chemistry of UTAD, Vila Real, from September to October 2010.References
- P. Gomez-Romero, C. Sanchez, C. Editors, Functional Hybrid Materials, 2003, Wiley Interscience, New York. Search PubMed.
- C. J. Brinker and G. W. Scherer, Sol–gel Science: The Physics and Chemistry of Sol–Gel Processing, 1990, Academic Press, San Diego, CA Search PubMed.
- C. Sanchez, B. Julián, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC.
- C. Sanchez, H. Arribart and M. M. Giraud Guille, Nat. Mater., 2005, 4, 277 CrossRef CAS.
- P. W. Anderson, Science, 1972, 177, 393 CrossRef CAS.
- N. Goldenfeld and L. P. Kadanoff, Science, 1999, 284, 87 Search PubMed.
- (a) R. B. Laughlin and D. Pines, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 28 CrossRef CAS; (b) J. Goldstein, Emergence, 1999, 1, 43 Search PubMed.
- S. Mann, S. L. Burkett, S. A. Davis, C. E. Fowler, N. H. Mendelson, S. D. Sims, D. Walsh and N. T. Whilton, Chem. Mater., 1997, 9, 2300 CrossRef CAS.
- (a) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartulli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS; (b) Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, P. M. Petroff, R. Leon, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317 CrossRef CAS; (c) Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. Guo, H. Soyez, B. Dunn, M. H. Huang and J. I. Zink, Nature, 1997, 389, 364 CrossRef CAS; (d) C. J. Brinker, Y. F. Lu, A. Sellinger and H. Y. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS; (e) S. L. Burkett, S. D. Sims and S. Mann, Chem. Commun., 1996, 1367 RSC; (f) S. Inagaki, S. Guan, T. Ohsuna and O. Terasaki, Nature, 2002, 416, 304 CrossRef CAS.
- J. J. E. Moreau, L. Vellutini, M. Wong Chi Man, C. Bied, J.-L. Bantignies, P. Dieudonne and J.-L. Sauvajol, J. Am. Chem. Soc., 2001, 123, 7957 CrossRef CAS.
- (a) J. J. E. Moreau, L. Vellutini, M. Wong Chi Man and C. Bied, Chem.–Eur. J., 2003, 9, 1594 CrossRef CAS; (b) J. J. E. Moreau, B. P. Pichon, M. Wong Chi Man, C. Bied, H. Pritzkow, J.-L. Bantignies, P. Dieudonné and J.-L. Sauvajol, Angew. Chem., Int. Ed., 2004, 43, 203 CrossRef CAS; (c) J. J. E. Moreau, L. Vellutini, M. Wong Chi Man, C. Bied, J.-L. Bantignies and J.-L. Sauvajol, Chem.–Eur. J., 2005, 11, 1527 CrossRef CAS.
- N. Liu, K. Yu, B. Smarsly, D. R. Dunphy, Y.-B. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2002, 124, 14540 CrossRef CAS.
- (a) Y. Fujimoto, A. Shimojima and K. Kuroda, Chem. Mater., 2003, 15, 4768 CrossRef CAS; (b) A. Shimojima and K. Kuroda, Angew. Chem., Int. Ed., 2003, 42, 4057 CrossRef CAS.
- J. J. E. Moreau, L. Vellutini, M. W. Chi Man and C. Bied, J. Am. Chem. Soc., 2001, 123, 1509 CrossRef CAS.
- J. Alauzun, A. Mehdi, C. Reye and R. J. P. Corriu, J. Mater. Chem., 2005, 15, 841 RSC.
- H. Tang, J. Sun, J. Jiang, X. Zhou, T. Hu, P. Xie and R. Zhang, J. Am. Chem. Soc., 2002, 124, 10482 CrossRef CAS.
- (a) B. Boury and R. J. P. Corriu, Chem. Commun., 2002, 795 RSC; (b) B. Boury and R. Corriu, Chem. Rev., 2003, 3, 120 Search PubMed.
- A. N. Parikh, M. A. Schivley, E. Koo, K. Seshadri, D. Aurentz, K. Mueller and D. L. Allara, J. Am. Chem. Soc., 1997, 119, 3135 CrossRef CAS.
- A. Shimojima, Y. Sugahara and K. Kuroda, Bull. Chem. Soc. Jpn., 1997, 70, 2847 CAS.
- (a) Y. Nakazaki, K. Fujita, K. Tanaka and T. Uchino, J. Phys. Chem. C, 2008, 112, 10878 Search PubMed; (b) A. Nishimura, N. Sagawa and T. Uchino, J. Phys. Chem. C, 2009, 113, 4260 Search PubMed; (c) A. Nishimura, S. Harada and T. Uchino, J. Phys. Chem. C, 2010, 114, 8568 Search PubMed.
- (a) L. D. Carlos, V. de Zea Bermudez, V. S. Amaral, S. C. Nunes, N. J. O. Silva, R. A. Sá Ferreira, C. V. Santilli, D. Ostrovskii and J. Rocha, Adv. Mater., 2007, 19, 341 CrossRef CAS; (b) S. C. Nunes, PhD. Dissertation, 2007, Vila Real (Portugal), 92678/1/2 SD, http://opac.sde.utad.pt/geral/ Search PubMed; (c) L. D. Carlos, J. M. Pacheco, R. A. Sá Ferreira and A. L. L. Videira, Small, 2010, 6, 386 Search PubMed.
- (a) S. C. Nunes, V. de Bermudez, J. Cybinska, R. A. Sá Ferreira, L. D. Carlos, M. M. Silva, M. J. Smith, D. Ostrovskii and J. Rocha, J. Mater. Chem., 2005, 15, 3876 RSC; (b) C. Anrez and A. Giannis, Eur. J. Org. Chem., 2001, 1, 137 Search PubMed; (c) S. Stevelmans, J. C. M. van Hest, J. F. G. A. Jansen, D. A. F. J. van Boxtel, E. M. M. de Brabander-van den Berg and E. W. Meijer, J. Am. Chem. Soc., 1996, 118, 7398 CrossRef CAS.
- M. Y. Hikosaka, S. H. Pulcinelli, C. V. Santilli, K. Dahmouche and A. F. Craievich, J. Non-Cryst. Solids, 2006, 352, 3705 CrossRef CAS.
- H. P. Klug and L. E. Alexander, X-Ray Diffraction Procedures for Polycrystalline and Amorphous Materials, 2nd Ed., Wiley, New York, 1974, ch. 9 Search PubMed.
- I. Horcas, R. Fernandez, J. M. Gomez-Rodriguez, J. Colchero, J. Gomez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 13705 CrossRef CAS.
- Peakfit is a product of the Jandel Corporation, 2591 Rerner Boulevard, San Rafael, CA 94901, U.S.A.
- Y. Fan, M. Kobayashi and H. Kise, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1318 CrossRef CAS.
- M. Pursch, A. Jäger, T. Schneller, R. Brindle, K. Albert and E. Lindner, Chem. Mater., 1996, 8, 1245 CrossRef CAS.
- (a) J. Clauss, K. Schmidt-Rohr, A. Adam, C. Boeffel and H. W. Spiess, Macromolecules, 1992, 25, 5208 CrossRef CAS; (b) L.-Q. Wang, J. Liu, G. J. Exarhos, K. Y. Flanigan and R. Bordia, J. Phys. Chem. B, 2000, 104, 2810 CrossRef CAS.
- L. D. Carlos, V. de Zea Bermudez, R. A. Sá Ferreira, L. Marques and M. Assunção, Chem. Mater., 1999, 11(3), 581 CrossRef CAS.
- C. Bohm, F. Leveiller, D. Jacquemain, H. Mohwald, K. Kjaer, J. Als-Nielsen, I. Weissbuch and L. Leiserowitz, Langmuir, 1994, 10, 830 CrossRef.
- (a) L. Fu, R. A. Sá Ferreira, N. J. O. Silva, L. D. Carlos, V. de Zea Bermudez and J. Rocha, Chem. Mater., 2004, 16(8), 1507 CrossRef CAS; (b) K. Dakmouche, C. V. Santilli, S. H. Pulcinelli and A. F. Craievich, J. Phys. Chem. B, 1999, 103, 4937 CrossRef CAS.
- J. F. Nagle and M. Goldstein, Macromolecules, 1985, 18, 2643 Search PubMed.
- (a) P. B. Braun, Nature, 1952, 170, 1123 CrossRef CAS; (b) G. W. Oosterhout and C. J. M. Rooymans, Nature, 1958, 181, 44 Search PubMed.
- S. Singh, J. Wegmann, K. Albert and K. Muller, J. Phys. Chem. B, 2002, 106, 878 CrossRef CAS.
- (a) R. A. Macphail, H. L. Strauss, R. G. Snyder and C. A. Ellinger, J. Phys. Chem., 1984, 88, 334 CrossRef CAS; (b) R. G. Snyder, H. L. Strauss and C. A. Ellinger, J. Phys. Chem., 1982, 86, 5145 CrossRef CAS.
- (a) N. V. Venkataram and S. Vasudevan, J. Phys. Chem. B, 2001, 105, 7639 CrossRef CAS; (b) N. V. Venkataram, S. Bhagyalakshmi, S. Vasudevan and R. Seshachi, Phys. Chem. Chem. Phys., 2002, 4, 4533 RSC.
- R. Wang, G. Baran and S. L. Wunder, Langmuir, 2000, 16, 6298 CrossRef CAS.
- K. G. Brown, E. Bicknell-Brown and M. Ladjadj, J. Phys. Chem., 1987, 91, 3436 CrossRef CAS.
- F. Bensebaa, Y. Zhou, A. G. Broto, D. E. Irish, Y. Deslandes, E. Kruss and T. H. Ellis, Spectrochim. Acta, Part A, 1999, 55, 1229 Search PubMed.
- D. J. Skrovanek, P. C. Painter and M. M. Coleman, Macromolecules, 1986, 19, 699 CrossRef CAS.
- D. J. Skrovanek, S. E. Howe, P. C. Painter and M. M. Coleman, Macromolecules, 1985, 18, 1676 CrossRef CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149 CrossRef CAS.
- L. D. Carlos, R. A. Sá Ferreira, V. de Zea Bermudez and S. J. L. Ribeiro, Adv. Funct. Mater., 2001, 11, 111 CrossRef CAS.
- L. D. Carlos, R. A. Sá Ferreira, R. N. Pereira, M. Assunção and V. de Zea Bermudez, J. Phys. Chem. B, 2004, 108, 14924 CrossRef CAS.
- S. S. Nobre, X. Cattoën, R. A. Sá Ferrerira, S. C. Carcel, V. de Zea Bermudez, M. W. Chi Man and L. D. Carlos, Chem. Mater., 2010, 22(12), 3599 CrossRef CAS.
- S. S. Nobre, C. D. S. Brites, R. A. Sá Ferreira, V. de Zea Bermudez, C. Carcel, J. J. E. Moreau, J. Rocha, M. Wong Chi Man and L. D. Carlos, J. Mater. Chem., 2008, 18, 4172 RSC.
- M. Fernandes, X. Cattoën, V. de Zea Bermudez and M. Wong Chi Man, CrystEngComm, 2011, 13, 1410 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis and characterization of m-A(x) and AC-m-A(8) mono-amidosils. See DOI: 10.1039/c2ra00702a |
| This journal is © The Royal Society of Chemistry 2012 |