A comparative study of air-dry and water swollen flax and cotton fibres
Sergey V.
Mikhalovsky
*a,
Vladimir M.
Gun'ko
ab,
Vladimir A.
Bershtein
c,
Vladimir V.
Turov
b,
Larisa M.
Egorova
c,
Claudine
Morvan
d and
Lyuba I.
Mikhalovska
a
aSchool of Pharmacy & Biomolecular Sciences, University of Brighton, Lewes Road, Brighton BN2 4GJ, UK. E-mail: s.mikhalovsky@brighton.ac.uk; Fax: +44 (0)1273642115; Tel: +44 (0)1273 642034
bChuiko Institute of Surface Chemistry, 17 General Naumov Str., Kiev, Ukraine. E-mail: vlad.gunko@ukr.net; Fax: +380 44 4243567; Tel: +380 44 4229627
cIoffe Physical-Technical Institute RAS, 194021 St-Petersburg, Russia. E-mail: vbersht.polmater@mail.ioffe.ru
dUniversite de Rouen/CNRS, UFR des Sciences, 76821 Mon-Saint-Aignan Cedex, France. E-mail: claudine.morvan@univ-rouen.fr
First published on 10th February 2012
Abstract
Thermal stability and structural characteristics of air-dry and swollen crude flax, bleached flax and cotton fibres and the behaviour of bound water were analysed using thermogravimetry, microscopy, differential scanning calorimetry, low-temperature 1H NMR spectroscopy and cryoporometry methods. Both air-dry and swollen fibres contain strongly (SBW) and weakly (WBW) bound water which differ in their behaviour at temperatures below 273 K. All samples have a higher content of SBW than WBW because of the structural features of natural fibres. The air-dry fibres studied have low porosity and similar inner mesoporous structure. The air-dry fibre samples of both cotton and bleached flax contain mainly SBW located in nanopores with radius R < 1–2 nm. In addition to SBW, the swollen fibres contain a significant fraction of WBW, which is located in mesopores with 1 < R < 10 nm, and practically unbound bulk water in large mesopores at R > 10 nm. According to cryoporometry, swelling substantially increases the pore volume (by a factor of 20–30) and specific surface area Smeso (two–three times) of fibres in the mesoporous region. The largest changes were observed in cotton fibres, owing to their chemical structure and textural characteristics affected by swelling. In the nanopore range, swelling reduced the specific surface area of nanopores (Snano) in cotton fibres and increased Snano in flax fibres, so that for air-dry samples of all fibres Snano > Smeso but for swollen samples Snano < Smeso.
Introduction
Natural plant fibres such as cotton and flax are typical soft materials widely used in textile industry, medical and technical applications.1 Of particular importance is the understanding of their interactions with water, as water strongly influences the properties and performance of fibres and fibre-based materials which can undergo substantial swelling. Cotton and flax fibres have been studied by many authors.2 However the behaviour and state of water in these materials and its effect on their inner porous structure merits further research.Cotton and flax fibres consist of elongated cells with average length of 2–4 cm. Their walls are thickened by apposition of so-called secondary walls (CW-II thickness ≤ 2 μm) with a large content of crystalline cellulose. There is a morphological and structural difference between cotton and flax fibres. Cotton fibres originate from the seed capsules as elementary trichome cells which adopt a so-called kidney shape during drying. Flax fibres develop in the stems between the cortex and the wood, within bundles where they are linked together by their middle lamellae and tricellular junctions. There are two specific structural properties of flax CW-II: it is composed of cellulose (80–90%) and a multilayer composite with cellulosic fibrils embedded in hemicellulose (up to 7%), pectins (up to 5%) and proteins (0–1.5%). A few phenolics (< 1%), waxes and fats (0.5–1.0%) and minerals (1%) are also present. In the main middle part of CW-II, the orientation of cellulose microfibrils almost follows the longitudinal axis of fibres.1–3 Cellulose fibrils aligned along the fibre length provide tensile and flexural strength and rigidity of fibres. The textural and structural organisation of natural fibres creates certain inner porosity as voids between adjacent cellulosic fibrils or in amorphous fragments.3,4 This porosity can be changed due to interaction with water, especially during fibre swelling. As a result, the adsorption capacity of fibres can be very different in dry and hydrated states of the materials.
Cotton and flax fibres can absorb relatively large amounts of moisture that can lead to microbial attack under certain conditions of humidity and temperature.5,6 Cotton and flax may act as a nutrient, becoming suitable medium for bacterial and fungal growth.5 Therefore, cotton and flax fibres are treated with numerous chemicals to get better antimicrobial textiles.6 Additionally, many of the physicochemical properties of natural fibres and composites based on fibres depend on the amount of bound water, adsorption capacity in respect to water and possible swelling degree.1,7 Thus, from practical and theoretical points of view, it is important to study the behaviour of water in air-dry and swollen flax and cotton fibres. Therefore, the aim of this paper is to compare the properties of air-dry and wetted/swollen flax and cotton fibres using differential scanning calorimetry (DSC), 1H NMR spectroscopy over a broad temperature range, and cryoporometry based on the NMR and DSC data.
Experimental
Materials
Crude and bleached cottonised flax fibres were prepared from the side product of flax scutching (Makarov Lenzavod, Kiev region, Ukraine), i.e. short fibres were carded in order to divide the technical fibres still linked by their tri-cellular junctions into elementary fibres. Non-bleached fibres were used in thermogravimetric (TG) and DSC measurements. In this paper the crude and bleached flax fibres are designated as flax and bleached flax, respectively. Commercial carded cotton fibres were obtained from Fisher (Cotton Absorbent, BP quality, cat. no. CTC-230-010S). Both the cottonised flax fibres and cotton fibres studied are used in bandages and wound care as flax wool and cotton wool, respectively.Characterisation methods
The freezing point of water in narrow pores at temperatures below 273 K can be described by the Gibbs–Thomson equation for the freezing point depression:9,10
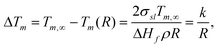 | (1) |
Results and discussion
SEM images of flax and cotton fibres (Fig. 1a and 1b) demonstrate that both flax and cotton wool have fibres of comparable diameter values (10–30 μm). However, their surface roughness is different, reflecting the different origin of the fibres (Fig. 1c and 1d) and preparation techniques. TEM images reveal the presence of some inner slit-shaped macropores (Fig. 1e and 1f), however, the fibre cell body is only weakly porous. | ||
| Fig. 1 (a–d) SEM and (e, f) TEM images of (a, c, e) bleached flax and (b, d, f) cotton fibres (scale bar (a, b) 20 μm and (c–f) 1 μm). | ||
Information on the interaction of fibres with water absorbed from air has been obtained from TG measurements. Weight loss vs. temperature is shown in Fig. 2. Cotton fibres absorbed 4.2 wt.% water, whereas both types of flax fibres absorbed 5.7 wt.% of water from air at 40% RH. The absorbed water is mainly lost upon heating below 373 K because the fibres are not nanoporous materials. The TG data show no considerable difference in the thermal behaviour of all three materials. Thermal destruction of the fibres started in the same temperature range of 500–510 K, with the main weight loss observed at 570–600 K (Fig. 2) due to decomposition/oxidation of cellulose and other organic components. More detailed information about the state of water retained by fibres has been obtained with DSC. Differential scanning calorimetry has been used to determine the amount and state of water bound in polymers and polymer membranes.11,12
 | ||
| Fig. 2 Thermogravimetric analysis of fibre samples (storage in air at RH = 40%). a–TG, b–DTG, b insert–enlarged fragment of DTG curves in the range of 300–380 K. | ||
This analysis is based on measuring the temperature, magnitude and shape of the melting endotherms (unfreezing of mobility) or the exotherms of ordering (crystallisation or quasi-crystallisation) of sorbed water. The endotherms of polymer dehydration have also been measured. The phase transitions in sorbed water have been observed in hydrophobic polymers, where the water formed a separate phase as droplets or clusters of micro- or submicro-volumes in cavities.11,12
The state of sorbed water in hydrophilic polymers is more complex. It has been found to exist in three states. Water I is the same as ‘normal’ bulk water, and its phase transitions are characterised by relatively narrow endothermic (melting) and exothermic (crystallisation) peaks on DSC curves; the temperature of these phase transitions is typically close to 273 K for ice-bulk water. It can be shifted at a high rate of cooling or heating. Notice that the 1H NMR signals of this water are not observed at T < 273 K since only mobile water is registered.8,9 The NMR cryoporometry method based on measuring the proton magnetic resonance signal of unfrozen water inside pores cannot detect water I at T < 273 K.
Water II is weakly bound but still capable of freezing by undergoing some ordering, structural reorganisation and quasi-crystallisation upon cooling, and disordering, or “melting” upon thawing at temperatures below 273 K.
Water III is strongly bound to polar groups of polymers. It does not show crystallisation or melting peaks on DSC curves. Water III forms a monomolecular water layer or a few monolayers (with a clustered structure) attached to macromolecules. The relative contributions of the three types of sorbed water depend on a number of factors such as the size of submicrocavities in the studied materials (inner pores in fibres) and molecular packing (free volume as voids between fibres). This is of particular importance for polymer membranes, the permeability of which is probably controlled mainly by water I.12 The thermal behaviour of waters II and III is the base of both DSC and NMR cryoporometry.8,9 However, the quantitative assignment of bound water to the mentioned three types on the basis of DSC and NMR methods can give slightly different contributions because of different sensitivity and different physical phenomena used in these methods.
Water III content may characterise to a certain extent the value of water/macromolecule surface contact. All three types of water are removed during dehydration of heated samples. Combined DSC, TG and NMR cryoporometry approaches allowed us to reveal the differences in the state of water sorbed by the fibres studied and to estimate the strength of water–fibre interactions and structural features of bound water and fibres. The dehydration enthalpy ΔHd values (Table 1) were determined from the area of fibre dehydration endotherms in the DSC curves. These values were compared with the standard value of specific enthalpy of “free” water evaporation, ΔH0vap. The ΔHd value is a complex parameter which includes the enthalpy required for breaking down water–fibre hydrogen bond interactions, the enthalpy of water diffusion within a fibre, and a ΔHvap contribution. The ΔHd/ΔHvap ratio characterises to a certain extent the relative strength of water–fibre interactions. The exothermic peaks of crystallisation and endothermic melting peaks of sorbed water were identified in two cases (Fig. 3, hatched areas, swollen flax and swollen cotton). The melting enthalpy ΔHm values were determined and compared with ice melting enthalpy ΔH0m. The ΔHm/ΔH0m ratio characterises the fraction of water which was converted into ice crystals. In swollen cotton and swollen flax only a small part of sorbed water (15.3% and 6.8%) are ice crystals at low temperatures (Table 1, ΔHm/ΔHm0). The other samples do not contain ice crystals at all. The ΔHm/ΔH0m ratio characterises the fraction of sorbed water I and partly water II because water III does not have a crystallisation peak in the thermograms (Fig. 3).
 | ||
| Fig. 3 DSC curves of fibre samples obtained below room temperature at cooling (on the left) or heating (on the right) with the rate of 5 K min−1. Curves 1–3 are for air-dry samples stored in air at 40% RH and curves 4–6 are for swollen samples after incubation for 24 h in distilled water. 1, 4–bleached flax; 2, 5–flax; 3, 6–cotton. | ||
| Sample | State | C w (wt%) | ΔHd (kJ g−1 water) | ΔHd/ΔHvap | ΔHm (J g−1 ice) | ΔHm/ΔH0m (%) |
|---|---|---|---|---|---|---|
| a ΔH0vap of water = 2.257 kJ g−1; ΔH0m ice = 334 J g−1. b n.d.–not detected. | ||||||
| Cotton | Air-dry, 40% RH | 4.2 | 11.74 | 5.20 | n.d.b | 0 |
| Swollen for 24 h | 26 | 6.15 | 2.72 | 51.1 | 15.3 | |
| Bleached flax | Air-dry, 40% RH | 5.7 | 6.30 | 2.78 | n.d. | 0 |
| Swollen for 24 h | 10 | 5.54 | 2.45 | n.d. | 0 | |
| Flax | Air-dry, 40% RH | 5.7 | 3.54 | 1.57 | n.d. | 0 |
| Swollen for 24 h | 17 | 5.32 | 2.35 | 22.9 | 6.8 | |
DSC measurements in a high sensitivity mode recorded a big difference in heat capacities ΔCp between swollen samples (Fig. 3, curves 4–6) in the low temperature region. The DSC curve showed no peaks for swollen bleached flax at a relatively low content of water (10 wt.%). For two other swollen samples the DSC curves revealed distinctive endo- and exothermic effects (Fig. 3, curves 5 and 6). For swollen flax (Cw = 17 wt.%), a small exothermic effect at 250–290 K upon cooling and the same endothermic effect at 235–268 K upon heating, with maxima at ∼240 and 260 K, were observed. It means that not only water III but also weakly bound water II is present in this sample accounting for about 6.8% of the total quantity of sorbed water. In the swollen cotton (Cw = 26 wt.%) all three types of sorbed water were present: sharp peaks of water crystallisation (Tmax = 261 K) and melting (Tmax = 276 K) indicate the presence of clusters of mobile water I; small exothermic peaks at 226 and 246 K are related to water II (Fig. 3). Water I and water II account for 15.3% of its total amount in the swollen cotton (Table 1). The rest of water in this material corresponds to non-crystallising water III, i.e. its structure is strongly distorted due to interactions with fibres. No such effects were found in the low temperature DSC curves for air-dry samples (Fig. 3, curves 1–3). It means that practically all water (∼4–6 wt.%) absorbed by air-dry fibres was strongly bound water of type III.
Fig. 4 shows the DSC curves of the fibres analysed at elevated temperatures. The endothermic effects characterise the dehydration process. It starts at lower temperatures and Tmax values are lower for air-dry samples than those for swollen samples, suggesting that it is more difficult to dehydrate the swollen fibres than air-dry samples. Secondly, the unusual plateau in the endotherm was reproducibly observed for swollen cotton at 390–418 K (Fig. 4a), which corresponds to a practically constant dehydration rate in this temperature region. This may be associated with a compensating effect of a decreasing amount of sorbed water vs. temperature rise.
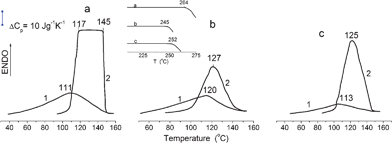 | ||
| Fig. 4 DSC dehydration endothermic curves of fibre samples obtained at elevated temperatures. (1) air-dry samples and (2) water swollen samples. Heating rate 20 K min−1. Insert: the exothermic bends in the DSC curves corresponding to the onset of cellulose destruction process in (a) cotton, (b) bleached flax and (c) flax. | ||
At elevated temperatures all air-dry samples have very different ΔHd values, which are significantly larger than ΔHvap values (Table 1). The ΔHd/ΔHvap ratios indicate that the “strength” of water III–fibre interactions in air-dry materials decreases in the order: cotton > bleached flax > flax. The differences in ΔHd and ΔHd/ΔHvap for swollen samples were much smaller. The insert in Fig. 4 shows the temperatures at which thermal destruction of fibre samples started. These temperatures are a little higher than values obtained in TG measurements, perhaps due to a higher heating rate in DSC experiments.
Additional information on the state of water in air-dry and swollen fibres was obtained using the low-temperature 1H NMR spectroscopy (Fig. 5 and 6) and cryoporometry (Fig. 7 and 8). The 1H NMR spectra (Fig. 5), amount of unfrozen water as a function of temperature (Cuw(T)) and the corresponding relationships between the Cuw(T) values and changes in the Gibbs free energy of water bound to fibres (Fig. 6) show that swollen cotton fibres contain a larger amount of both strongly bound water of type III (CSBW) and weakly bound water of type II (CWBW) than swollen bleached flax fibres (Table 2). This conclusion is in agreement with the DSC data despite the difference in swelling time. The air-dry bleached flax fibres contain larger amount of weakly bound water than air-dry cotton fibres.
 | ||
| Fig. 5 1H NMR spectra of air-dry (a, b) and 1 h-incubated with water (c, d) fibres recorded at different temperatures. (a, c) bleached flax, (b, d) cotton. | ||
 | ||
| Fig. 6 Amount of unfrozen water Cuw as a function of temperature for (a) air-dry and (b) swollen samples, and (c, d) corresponding relationships between Cuw and changes in the Gibbs free energy of bound water, ΔG. | ||
 | ||
| Fig. 7 Integral pore size distributions in (a) air-dry fibres and (b) fibres incubated with water for 1 h. | ||
 | ||
| Fig. 8 Pore size distributions for flax and cotton fibres calculated using DSC data (Fig. 3) and DSC-cryoporometry method. | ||
| Sample | C SBW (mg g−1) | C WBW (mg g−1) | ΔGs (kJ mol−1) | S uw (m2 g−1) | S nano (m2 g−1) | V nano (cm2 g−1) | S meso (m2 g−1) | V meso (cm3 g−1) | S BET,w 13 (m2 g−1) |
|---|---|---|---|---|---|---|---|---|---|
| Air-dry bleached flax | 28 | 10 | −2.94 | 10.9 | 6.0 | 0.003 | 4.8 | 0.032 | |
| Air-dry cotton | 33 | 6 | −3.00 | 19.2 | 14.0 | 0.007 | 5.1 | 0.031 | |
| Swollen bleached flax | 100 | 70 | −2.97 | 51.7 | 10.4 | 0.005 | 31.7 | 0.533 | 46 |
| Swollen cotton | 270 | 280 | −2.94 | 88.5 | 9.3 | 0.005 | 67.7 | 0.946 | 34 |
NMR cryoporometry8,14 was used to calculate the pore size distribution, PSD (Fig. 7) using the Cuw(T) functions (Fig. 6). Integration of PSD functions gives the textural characteristics of the samples (Table 2). The specific surface area (Suw = 51.7 m2 g−1, Table 2) of bleached flax fibres in contact with unfrozen bound water at 273 K is close to the specific surface area determined from the water sorption isotherm (SBET,w = 46 m2 g−1, Table 2).13
However, Suw ≫ SBET,w for 1 h incubated cotton. This difference in the bleached flax and cotton fibre properties can be explained by features in the fibre interactions with water (see above DSC results) and smaller amounts of water sorbed from the water vapour (0.15–0.2 g H2O per gram of fibre, but close to that used on the DSC measurement of swollen fibres) during measurements of the water sorption isotherms than during the NMR measurements (Fig. 6b).
The PSD (Fig. 7) and the textural characteristics (Table 2) of swollen fibres show that the contribution of mesopores (pore radius 1 < R < 25 nm) to the total surface area and especially to the total pore volume are significantly larger than that of nanopores (R < 1 nm). Compared to the air-dry samples, both Smeso and Vmeso increased substantially whereas changes in Snano and Vnano were minor (because the fibres studied are not nanoporous materials).
The increase in the S and V parameters is due to swelling since bound water enlarges the porosity of fibres, which is evident by comparing PSD curves for air-dry (Fig. 7a) and swollen (Fig. 7b) fibres, and their Vmeso values (Table 2). Notice that water was not removed from swollen samples used in the NMR measurements (Cuw = CSBW + CWBW < 0.55 g g−1) (Table 2) in contrast to the DSC measurements (Cw ≤ 0.26 g g−1). The melting temperature varies in a broad range for strongly bound water III which filled nanopores where R < 1 nm (Fig. 7, curves 3). Weakly bound water II is located in mesopores where R > 1 nm. Its melting occurs in a narrow temperature range close to 273 K (Fig. 6b). The major amount of water II in swollen fibres (Fig. 7b) is located in pores where R > 10 nm, and the influence of the surface is low. Therefore Tm for this type of water is close to 273 K. A portion of water located in mesopores (R < 25 nm) can be assigned to water I because the Vmeso value is larger than the volume occupied by SBW and WBW for both swollen cotton and swollen bleached flax (Table 2).
Calculations of the PSD for hydrated (swollen) flax and cotton fibres using the DSC-cryoporometry21 data (Fig. 3) give a picture (Fig. 8) which is in general agreement with the NMR-cryoporometry results (Fig. 7).
However, certain details of the NMR and DSC PSD differ because of different conditions of hydration (1 and 24 h), different amounts of water in the samples and different nature of these cryoporometry methods.8–10,15 The narrowest DSC PSD peaks at R ≈ 1.4 nm (Fig. 8) are shifted towards larger R values in comparison with the NMR PSD at R = 1 nm (Fig. 7b) because of the longer swelling time before the DSC measurements that can result in larger nanopore sizes. Intensity of the DSC PSD is lower for flax than cotton (Fig. 8) because of a lower content of water (17 and 26 wt.% respectively). The DSC measurements for bleached flax did not produce curves (Fig. 3) suitable for calculations of the PSD due to a low content of bound water (10 wt.%).
Conclusions
Flax and cotton fibres have comparable thermal stability, high hydrophilicity and swelling ability but different distributions of states of water sorbed on air-dry and wetted samples. The air-dry fibre samples of both cotton and bleached flax contain mainly strongly bound water (type III) located in nanopores with radius R < 1–2 nm. In addition to water type III, the swollen cotton and swollen bleached flax fibres contain a significant fraction of weakly bound water (type II), which is located in mesopores with 1 < R < 10 nm, and practically unbound bulk water in large mesopores at R > 10 nm. According to the NMR cryoporometry data, even short swelling of fibres in water significantly changes the pore size distribution in bleached flax and cotton fibres substantially increasing mesopore volume and surface area. The results obtained provide a deeper insight into features of sorption of different compounds on air-dry and swollen natural fibres such as cotton and flax.Acknowledgements
The work was supported by the Interreg IVA (South) project 4044 Flax and FP7-PEOPLE-IRSES project 230790 COMPOSITUM.References
- (a) K. G. Satyanarayana, G. G. C. Arizaga and F. Wypych, Prog. Polym. Sci., 2009, 34, 982–1021 CrossRef CAS; (b) M. J. John and S. Thomas, Carbohydr. Polym., 2008, 71, 343–364 CrossRef CAS; (c) S. Alix, S. Marais, C. Morvan and L. Lebrun, Composites, Part A, 2008, 39, 1793–1801 CrossRef; (d) S. Alix, E. Philippe, A. Bessadok, L. Lebrun, C. Morvan and S. Marais, Bioresour. Technol., 2009, 100, 4742–4749 CrossRef CAS.
- (a) B. Madsen, A. Thygesen and H. Lilholt, Compos. Sci. Technol., 2009, 69, 1057–1069 CrossRef CAS; (b) N. E. Zafeiropoulos, C. A. Baillie and F. L. Matthews, Composites, Part A, 2001, 32, 525–543 CrossRef; (c) T. T. Teeri, H. Brumer III, G. Daniel and P. Gatenholm, Trends Biotechnol., 2007, 25, 299–306 CrossRef CAS; (d) J. Andersons, E. Spãrņiš, R. Joffe and L. Wallström, Compos. Sci. Technol., 2004, 65, 693–702 CrossRef; (e) G. Cantero, A. Arbelaiz, R. Llano-Ponte and I. Mondragon, Compos. Sci. Technol., 2003, 63, 1247–1254 CrossRef CAS; (f) K. Charlet, C. Baley, C. Morvan, J. P. Jernot, M. Gomina and J. Bréard, Composites, Part A, 2007, 38, 1912–1921 CrossRef.
- P. Zugenmaier, Crystalline Cellulose and Cellulose Derivatives. Characterization and Structures, Springer-Verlag, Berlin, 2008 Search PubMed.
- M. R. Rowell, R. A. Young and J. K. Rowell (ed.) Paper and Composites from Agro-based Resources, CRC Lewis Publishers, New York, 1997 Search PubMed.
- (a) Y. Gao and R. Cranston, Text. Res. J., 2008, 78, 60–72 CrossRef CAS; (b) M. Gorenňsek and P. Recelj, Text. Res. J., 2007, 77, 138–141 CrossRef; (c) S. Ravindra, Y. M. Mohan, N. N. Reddy and K. M. Raju, Colloids Surf., A, 2010, 367, 31–40 CrossRef CAS.
- (a) Y. A. Son, B. S. Kim, K. Ravikumar and S. G. Lee, Eur. Polym. J., 2006, 42, 3059–3067 CrossRef CAS; (b) E. Falletta, M. Bonini, E. Fratini, A. Lo Nostro, A. Becheri, P. Lo Nostro and P. Baglioni, Int. J. Nanotechnol., 2007, 4, 412–414 CrossRef CAS; (c) W. K. Son, J. H. Youk and W. H. Park, Carbohydr. Polym., 2006, 65, 430–434 CrossRef CAS; (d) S. Tarimala, N. Kothari, N. Abidi, E. Hequet, J. Fralick and L. L. Dai, J. Appl. Polym. Sci., 2006, 101, 2938–2943 CrossRef CAS.
- (a) A. Le Duigou, P. Davies and C. Baley, Compos. Sci. Technol., 2010, 70, 231–239 CrossRef CAS; (b) N. Sgriccia and M. C. Hawley, Compos. Sci. Technol., 2007, 67, 1986–1991 CrossRef CAS; (c) Y. Xie, C. A. S. Hill, Z. Xiao, H. Militz and C. Mai, Composites, Part A, 2010, 41, 806–819 CrossRef; (d) J. Bras, M. L. Hassan, C. Bruzesse, E. A. Hassan, N. A. El-Wakil and A. Dufresne, Ind. Crops Prod., 2010, 32, 627–633 CrossRef CAS.
- V. M. Gun'ko, V. V. Turov, R. Leboda, V. I. Zarko, J. Skubiszewska-Zięba and B. Charmas, Langmuir, 2007, 23, 3184–3192 CrossRef CAS.
- J. Mitchell, J. B. W. Webber and J. H. Strange, Phys. Rep., 2008, 461, 1–36 CrossRef CAS.
- (a) D. W. Aksnes, K. Forland and L. Kimtys, Phys. Chem. Chem. Phys., 2001, 3, 3203–3207 RSC; (b) J. H. Strange, M. Rahman and E. G. Smith, Phys. Rev. Lett., 1993, 71, 3589–3591 CrossRef CAS.
- S. Rowland, Water in Polymers, Amer. Chem. Soc., Washington, D.C. 1980 Search PubMed.
- V. A. Bershtein and V. M. Egorov, Differential Scanning Calorimetry of Polymers. Physics, Chemistry, Analysis, Technology, Ellis Horwood, New York, 1994 Search PubMed.
- L. I. Mikhalovska, V. M. Gun'ko, A. A. Rugal, O. I. Oranska, Y. I. Gornikov, C. Morvan, C. Domas and S. V. Mikhalovsky, RSC Advances, 2012 10.1039/C2RA00725H.
- V. M. Gun'ko, V. V. Turov, V. M. Bogatyrev, V. I. Zarko, R. Leboda, E. V. Goncharuk, A. A. Novza, A. V. Turov and A. A. Chuiko, Adv. Colloid Interface Sci., 2005, 118, 125–172 CAS.
- (a) M. R. Landry, Thermochim. Acta, 2005, 433, 27 CrossRef CAS; (b) G. Rohman, F. Lauprêtre, S. Boileau, P. Guérin and D. Grande, Polymer, 2007, 48, 7017 CrossRef CAS; (c) J. Weber and L. Bergström, Langmuir, 2010, 26, 10158 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |