Monosaccharide derivatives as central scaffolds in the synthesis of glycosylated drugs
Marco
Filice
* and
Jose M.
Palomo
*
Departamento de Biocatálisis. Instituto de Catálisis (CSIC) c/Marie curie 2, Cantoblanco, campus UAM, 28049, Madrid, Spain. E-mail: marcof@icp.csic.es; josempalomo@icp.csic.es
First published on 3rd January 2012
Abstract
Carbohydrates are key molecules in nature with multiple roles in biological processes. Specifically the biological function of the glycosylated part is of great importance in natural products. In many cases carbohydrate-containing molecules are structurally diverse and this heterogeneity makes the isolation of sufficient amounts of the organic molecule from biological source extremely difficult. Therefore, chemical synthesis offers the advantage of producing pure and structurally defined oligosaccharides for glycoconjugate synthesis for biological investigations. Although the synthesis of these glycoderivatives is hampered by difficulties associated with the regioselectivity in polyhydroxyl protection and the stereoselectivity of glycosidic linkages, the preparation of activated regioselective sugar units, could represent a huge challenge in glycochemistry. The present review discusses some of the most recent advances in the construction of a set of tailor-made monosaccharide derivatives, key tools for final glycosylated drugs development.
 Marco Filice | Marco Filice obtained his BS in pharmaceutical chemistry and technology at the University of Pavia, Italy, in 2003. He received his Ph.D. degree (summa cum laude) in 2007 at the Pharmaceutical Chemistry Department in the University of Pavia. For postdoctoral research he joined to the group of Prof. Guisan at Biocatalysis Department in Institute of Catalysis (IPC, CSIC) in Madrid in 2008 where currently he works as an Associate Research Scientist with a JAE-DOC contract. His current research interests include carbohydrate and nucleoside chemistry with enzymes, applied biocatalysis, site-directed chemical modification of enzymes and asymmetric biotransformations in fine and pharmaceutical chemistry. |
 Jose M. Palomo | Jose M. Palomo received his Ph.D. degree (summa cum laude) in 2003 in the Autonoma University in Madrid working at the Biocatalysis Department in Institute of Catalysis (IPC, CSIC) in Madrid. For postdoctoral research he joined to the group of Prof. Waldmann at Chemical Biology Department of Max Planck Institute in Dortmund in 2004. In 2006, he began his appointment as an Associate Research Scientist at Biocatalysis Department ICP-CSIC. From 2009, he is a Tenured Scientist (Associate Professor) in ICP-CSIC. His current research interests include design of semisynthetic enzymes, asymmetric biotransformations and carbohydrate chemistry with enzymes. |
Introduction
Carbohydrates represent complex and structurally diverse molecules containing multiple chiral centers which are used in nature to synthesize useful complex bioactive compounds. They can exist as oligo- and polysaccharides or included in aglycon structures as glycoderivatives (peptides, lipids, etc.) with key roles in a broad range of biological processes including signal transduction, carcinogenesis and immune responses.1–5The preparation of these biomolecules is not straightforward—because of their complex structural diversity6–8—unlike other similar compounds such as nucleic acids or proteins, where no regio- and stereochemical issues are involved in the sequential coupling steps for the construction of phosphate or amide bonds, respectively.
In many cases the biological activity of natural products derives from the sugar moieties. Changes on the sugar structures cause deep impact on the biological properties of the parent compounds.9,10 Glycoderivatization of natural products by changing the sugar moiety on the molecule has been performed via chemical or biosynthetic pathways.11 Some interesting improvements in antitumoral activity was found in erythromycin,12 vancomycin,13 or lately digitoxin analogues.14
Furthermore, a way to enhance functional properties of drugs such as solubility, pharmacokinetic or pharmacodynamic consists of the incorporation of carbohydrate moieties to the molecules.15–17
For example, tilorone, 2,7-bis[2(diethylamino)-ethoxy]-9H-fluoren-9-one glycosylated analogues improved its property of induction IFN production which, in turn, might stimulate cytostatic effects in the cell in vivo against some cancers18 or biological activity, and the solubility of taxol was improved after a non-natural glycosylation.19
Glycosylated steroids have been shown to possess various types of biological activities. The glycosylation of steroids, analogues of cardenolides and saponins, enhanced the biological activity for their potential use in drug delivery systems.20 However, an established chemical approach for oligosaccharide or glycoderivative synthesis involved multi-step protection and glycosylation steps. Therefore, the development of strategies to achieve an efficient and easy glycosylated natural product to produce new powerful drugs is of great interest.
In this way, the accessibility of new building blocks by an efficient and simple way represents an outstanding challenge in glycochemistry. The preparation of selectively protected monosaccharide units bearing a single strategically positioned free hydroxyl group (a nucleophilic acceptor) symbolizes a breakthrough in carbohydrate synthesis (Scheme 1) together with the stereoselective glycosylation.21–26
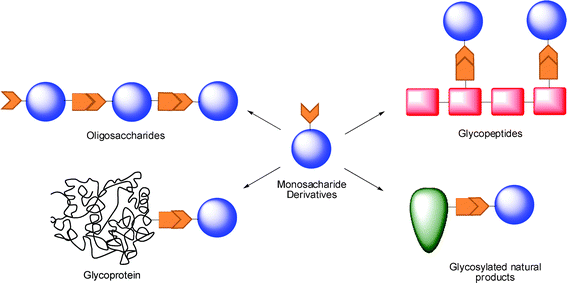 | ||
| Scheme 1 Tailor-made glycosylated molecule structures by this approach. | ||
In the present review, we present the most recent examples in the preparation of monosaccharide derivatives and their application as key scaffolds in the synthesis of different biologically active glycosylated drugs.
1. Chemical strategies to prepare monosaccharide derivatives for biologically active glycosylated drug synthesis
One of the most interesting strategies to prepare monohydroxy building blocks is based on the necessity to install suitable protecting groups on the remaining hydroxyls, with the aim of tuning the overall electronic properties of the donors and acceptors so as to ‘match’ the donor–acceptor pair and also for further deprotection and glycosylation or functional-group modifications.Hung and co-workers have described a combinatorial and highly regioselective novel method to protect individual hydroxyl groups of a monosaccharide in different positions which was quite useful for the synthesis of influenza virus oligosaccharides.27
This approach permits to install an orthogonal protecting group pattern in a ‘one-pot’ reaction by Lewis acid catalysis (trimethylsilyl trifluoromethansulfonate (TMSOTf) or Cu(OTf)2), by-passing isolation and purification of intermediates in the synthesis.
A correct choice of the protecting groups is crucial for the successful development of this strategy. Substituted and unsubstituted benzyl ethers were chosen. They act as orthogonal protecting groups and can be cleaved advantageously by means of reagent combination.28 Different protected monosaccharides, 2-alcohols, 3-alcohols, 4-alcohols, and 6-alcohols starting from 2,3,4,6-tetra-O-trimethylsilylated monosaccharides, were obtained by this methodology (Scheme 2).
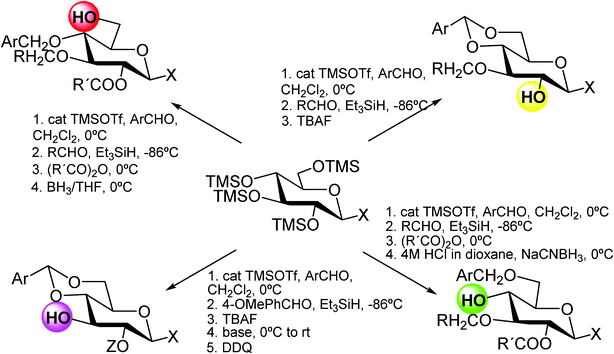 | ||
| Scheme 2 | ||
Hundreds of building blocks starting from D-glucose—with different aliphatic, aromatic substituents in anomeric position—have been efficiently prepared in high overall yields (Table 1).
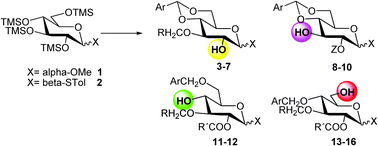
| Entry | X | Ar | Z | R | R′ | Product | Yield [%] |
|---|---|---|---|---|---|---|---|
| 1 | 1 | Ph | Ph | 3 | 91 | ||
| 2 | 1 | Ph | 4–OMePh | 4 | 94 | ||
| 3 | 1 | Ph | 3,4–diOMePh | 5 | 81 | ||
| 4 | 1 | 4–NO2Ph | 3,4–diOMePh | 6 | 81 | ||
| 5 | 1 | 2-naphthyl | Ph | 7 | 80 | ||
| 6 | 1 | Ph | Allyl | 8 | 73 | ||
| 7 | 2 | Ph | Bu | 9 | 65 | ||
| 8 | 2 | Ph | Allyl | 10 | 70 | ||
| 9 | 2 | Ph | Ph | CH3 | 11 | 70 | |
| 10 | 2 | Ph | 2-naphthyl | CH3 | 12 | 67 | |
| 11 | 1 | Ph | Ph | Ph | 13 | 65 | |
| 12 | 1 | Ph | 2-naphthyl | CH3 | 14 | 60 | |
| 13 | 1 | 2-naphthyl | 2-naphthyl | CH3 | 15 | 63 | |
| 14 | 1 | 2-naphthyl | 2-naphthyl | Ph | 16 | 65 |
Some of them were applied as scaffolds in the preparation of H5N1 avian influenza virus binding trisaccharide 22 and different analogues 23 and 24.27
The glycosylation reaction between the protected syalic acid thioderivative 17 and the monohydroxy building block 18 by a AgOTf mediated activation produced the disaccharide 19 (Scheme 3) which was followed by p-toluenylsulphenyl trifluoromethanesulphonate (p-TolSOTf)-promoted coupling with the alcohols 20, 13, and 21 in a one-pot manner, yielded the expected linear trisaccharides 22, 23, and 24, respectively.27
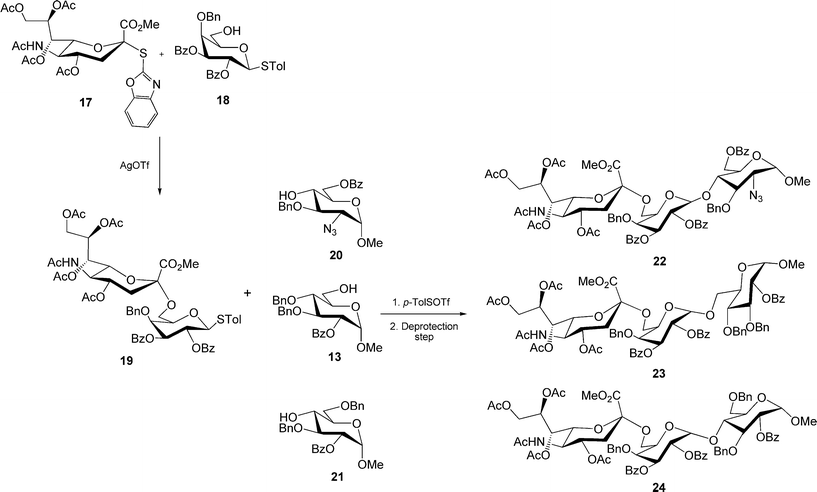 | ||
| Scheme 3 | ||
Furthermore, another interesting application of this straightforward strategy is represented by the construction of branched oligosaccharides (Scheme 4).
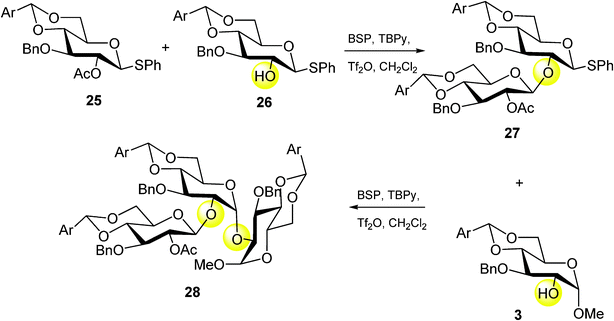 | ||
| Scheme 4 | ||
In this case, the utility of the one-pot syntheses of monomeric building blocks was applied to synthesize the protected trimeric oligosaccharide by an efficient iterative construction.27
Starting from the monomeric unit 26, a phenylsulfenyl-promoted assembly with 25 was performed providing exclusively β-dimeric glycosyl donor 27 in 70% yield. Further activation of 27 and coupling with the acceptor 3 yielded exclusively the trisaccharide 28 in 66% yield (Scheme 4).
Shao et al.29 describe the synthesis of 1,2-cyclopropaneacetylated protected monosaccharides and their use as a new class of glycosyl donor (Table 2).
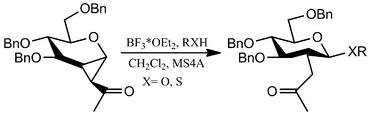
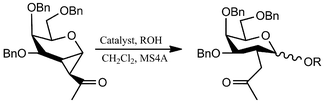



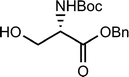
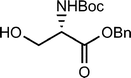
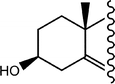

The glycosylation reaction was efficient and provides a method for the stereoselective synthesis of 2-C-acetylmethyl-2-deoxy-glycosides, oligosaccharides, glycosylamino acids, and other 2-C-acetylmethylglycosides with interesting applications in the preparation of glucosidase inhibitors, HIV virus inhibitors, etc.
Despite the multistep pathway30 required for the obtainment of the cyclopropanated glucosyl and galactosyl donors, this Lewis-acid catalyzed reaction is noteworthy especially for the complete control of the stereoselectivity obtained through the right combination between the donor configuration (gluco- vs. galacto- structure) and the Lewis-acid catalyst employed. In fact, the stereoselectivity of the glucosyl donors glycosylation resulted completely in favor of the β-anomer products (independently from the added Lewis acid) from good to excellent yields (79–93%), whereas the stereoselectivity in couplings of a galactosyl donor with acceptors depended on the catalysts selected: BF3*OEt2 to predominantly obtain β-anomer and TMSOTf to predominantly obtain the α-anomer (Table 2).
Another very interesting research work is represented by the investigation of Than et al.31 By combining systematic geometrical diversity with systematic functional diversity,32 this research group reported the development of a versatile and practical multistep solid-phase synthetic route applied to two monosaccharide scaffolds (glucose 41 and allose 42 derivatives, even if it can be adapted to other pyranose building blocks) which allowed the generation of easy-to-evaluate libraries of pyranose based drug-like peptide mimetics (Scheme 5).
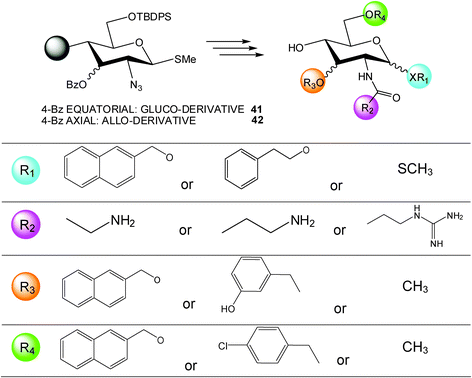 | ||
| Scheme 5 | ||
This methodology permits the introduction of a wide range of substituent families in a regio- and stereospecific manner, furthermore, several synthetical steps have been performed on solid phase (Wang resin) simplifying and improving the overall route. Certainly, the entire process is affected by laborious procedures but undoubtedly its great strength relies in the possibility to furnish an interesting general alternative in the creation of a vast pyranose based peptide mimetic library.
Once elaborated, the systematic structurally diverse library (planned around a tripeptide motif containing two aromatics and one positive charge) was screened against the somatostatin receptor subtypes (sst1-5) and the melanin-concentrating hormone receptor 1 (MCH1), examples of class A G-protein coupled receptors (GPCR) known to bind peptides containing this tripeptide motif and to directly impact to some physiological and pathological processes (i.e. modulation of cell proliferation, modulation of many endocrine and exocrine secretory processes, body weight regulation, etc.).33–38 The results were satisfactorily identifying, among hundreds of molecules produced in the library, a small set of molecules possessing a submicromolar IC50 against the receptors above mentioned.39
Yang et al.40 describe a unique and rapid approach towards the synthesis of triazolyl pseudo-glycopeptides by employing microwave-accelerated Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (CuAAC or ‘click’ reaction) (Scheme 6). By this way, a series of triazole-linked serinyl, threoninyl, phenylalaninyl and tyrosinyl perbenzylated-1-O-gluco- or galactosides 48–51 have been efficiently and rapidly synthesized in high yields.
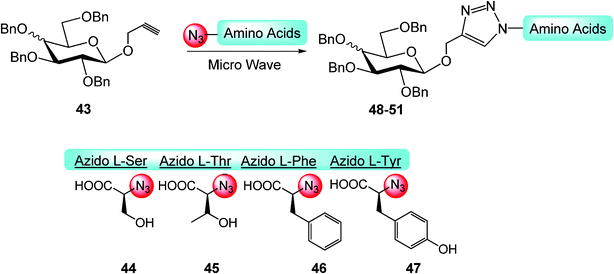 | ||
| Scheme 6 | ||
These glycopeptidotriazoles showed a favorable biological activity as inhibitors toward some elements of the protein tyrosine phosphatase (PTP), an enzymatic superfamily whose inappropriate regulation of many of its members has been reported to be causative toward the pathogenesis of major human diseases involving autoimmune disorders, diabetes and cancer.41
With this approach, by using inexpensive and abundant natural scaffolds (sugars and amino acids) and via facile and expeditious synthetic methods (microwave-assisted ‘click’ reaction), a pseudo-glycopeptide-based library has been efficiently produced, furnishing new perspectives toward the development of very active PTP inhibitors (Scheme 6).
Together with the synthesis of new bioactive molecules, the chemical modification of natural compounds with proven pharmacological activity (in order to improve their properties) ever resulted as a powerful strategy deeply investigated by many research groups. Following this promising line, for example, a wide range of bicyclic sugar-shaped analogs of the natural iminosugars castanospermine or nojirimycin have been developed by the Garcia-Fernandez group.42,43 The natural parental alkaloids possess a potential biological activity as competitive inhibitors of several glucosidases in the treatment of diseases such as cancer, viral infections, diabetes or glycosphingolipid storage disorders. However, their generally low selectivity together with the poor cell permeability (due to the highly hydrophilic nature) resulted in disappointing failures in most clinical trials.
To overcome these drawbacks, the Garcia-Fernandez research group developed a new family of glycomimetics, incorporating an endocyclic nitrogen atom (with a high sp2-hybridisation character: sp2 iminosugar) together with an hydrophobic modification.44
In this recent publication, an improved stereoselective synthesis of stable sp2-iminosugar-type N– (52), S– (53), and C– (54) glycoside castanospermine analogues bearing a α-configured pseudoanomeric group (Scheme 7) has been reported.44 The methodology involves a common pivotal intermediate and it is well suited for library generation. Biological activity of the new synthetic castanospermin analogues was tested resulting in low- to sub-micromolar inhibitors of neutral α-glucosidase exhibiting much higher enzyme selectivity when compared with the parent alkaloid castanospermine.
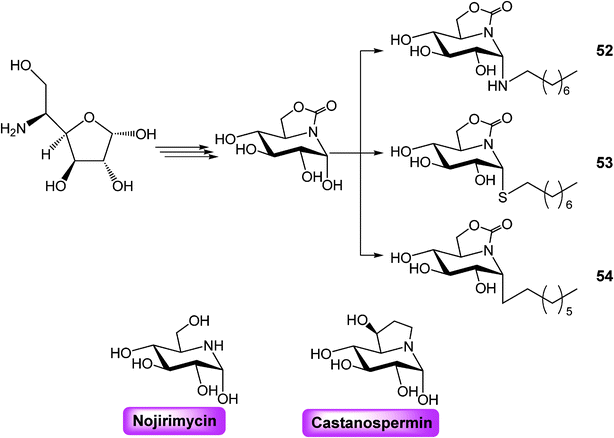 | ||
| Scheme 7 | ||
Over the past two decades, the Danishefsky research group has been notoriously dedicated to the design and de novo synthesis of carbohydrate and peptide-based antitumor vaccine constructs that, if properly presented to the immune system, could stimulate the formation of antibodies which would selectively bind and eradicate tumor cells overexpressing the carbohydrate epitopes at issue.
Specially, the main goal of this fascinating research area was centered on the development of immunostimulating strategies allowing for enhanced protection against tumor recurrence and metastasis following elimination of the primary tumor mass through the canonical protocols (i.e. surgery, radiation or chemotherapeutic treatment).
Firstly, this research was approached preparing monovalent vaccines (a single carbohydrate antigen conjugated to an immunogenic carrier molecule),45 subsequently, the second generation studies were focused on the preparation of more elaborated constructs: multiple repeats of a carbohydrate epitope (clusters) anchored on a peptide backbone.46 In order to amplify potency of a broader base, a third generation of carbohydrate-based antitumor vaccines were synthesized. These new constructs were elaborated keeping in mind the evidence that any single tumor cell is characterized by a significant heterogeneity of its surface in carbohydrate antigens expression. Based on this thesis, unimolecular multiantigenics vaccine constructs have been prepared.47
Recently, this research area underwent a further evolution by considering the possibility that the peptide backbone might also act not only as an antigenic linker protein but also like a potent additional antigenic marker. On the basis of these observations, the Danishefsky group designed a new type of antitumor vaccine structure featuring both a carbohydrate-based antigen and a mucin-derived peptide-based marker in an alternating reiterative pattern (Scheme 8).48
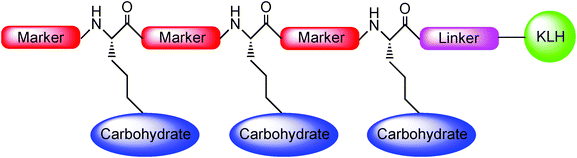 | ||
| Scheme 8 | ||
Recurring to a wide set of modern and straightforward synthetic procedures (glycopeptide solid phase synthesis, modified and optimized olefin cross metathesis, etc.), the Gb3-glycosylaminoacyl 55 building block has been prepared.48
Subsequently, cassette 55 was modified in order to install two N-termini bearing different compatible orthogonal protecting groups. So that, iterative peptide couplings with the MUC5AC-based peptide marker (a 8-amino acid epitope—sequence: TTSTTSAP—belonging to the peptidic mucin antigen MUC5AC expressed on the ovarian cancer cell surface and prepared through Fmoc solid-phase synthesis using Novabiochem proline-TGT resin) followed by selective deprotection of the desired N-termini permitted to achieve the fully synthetic clustered Gb3-MUC5AC construct.
Finally, bioconjugation with the immunogenic carrier KLH (in a 698![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) afforded the vaccine construct Gb3-MUC5AC cluster KLH conjugate 56 targeting the ovarian carcinoma (Scheme 9).
:1 ratio) afforded the vaccine construct Gb3-MUC5AC cluster KLH conjugate 56 targeting the ovarian carcinoma (Scheme 9).
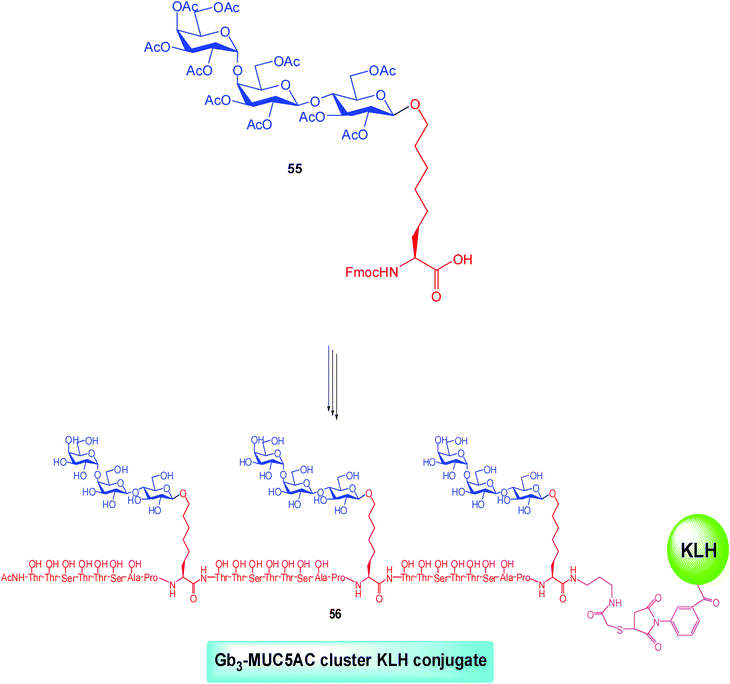 | ||
| Scheme 9 | ||
The yield of the final product was not very high (<10% for the entire synthetic pathway) but, in this case, the main purpose was to obtain a homogenous molecule useful for clinical trials. Hitherto, the clinical results of this last version of the carbohydrate vaccine have not yet published, but considering the good results obtained with the former versions of the vaccine constructs,49 it can be speculated that they could be noteworthy.
The chemical reaction set applied here demonstrates, once again, the awesome ability of the Danishefsky group handling the total synthesis in an effort to mimic nature. In fact, such specified structures have been produced through a rigid synthetical control otherwise almost impossible to reproduce only recurring to strictly biological procedures. So, total chemical synthesis can be clearly employed to reproduce or enhance the performance of molecules which had hitherto been considered exclusively as “biologics”.
2. Enzymatic strategies to prepare monosaccharide derivatives for chemoenzymatic glycosylated drug synthesis
A huge number of methods for the regioselective protection of monosaccharides has been reported in the literature, although the preparation of carbohydrate based building blocks through a regioselective deprotection of their fully protected precursors results has not been explored so far. In fact, this approach, especially in the case of the preparation of carbohydrate acceptors, may be rather unfeasible due to the difficulty in efficiently deprotecting a unique position—leaving the remaining ones protected—when employing the same hydroxyl chemical protecting group.Nevertheless, deprotection at primary or secondary positions, by the majority of reported chemical methods, does not completely fulfill the requirement of a very regioselective methodology. In fact, because of the difficulty to discriminate between groups with a very similar reactivity, the chemical deprotection is the limiting step especially when using monosacharides with all the hydroxyls blocked by a unique protecting group. By this way, a biocatalytic approach has represented a convenient alternative. Enzymes, in many cases, exhibit good catalytic activity, specificity and regioselectivity, transforming only a substrate in an unique product from different possible enantio- or regioisomers.
In this way, a nice part of work is based on the development of a combinatorial biocatalytic methodology to prepare—by one-pot synthesis starting from cheap fully acetylated molecules—a small library of different monohydroxy monosaccharides.50–53
The strategy is based on the complex catalytic mechanism of lipases which implies dramatic conformational changes of the enzyme molecule between a closed and an open form.54 Thus the application of different immobilization protocols for a particular lipase—involving different areas of the lipase, rigidity, micro-environments, etc.—has given access to different biocatalysts for the same enzyme, altering its activity and regioselectivity during the hydrolysis of peracetylated carbohydrates in aqueous media.
A case which perfectly exemplified the power of this strategy is represented by the regioselective biocatalytic hydrolysis of β-monosaccharides.50
For example, by this strategy, three different biocatalysts from Thermomyces lanuginose lipase (TLL) showed quite different catalytic properties in the hydrolysis of peracetylated β-galactose 57 (Scheme 10).51
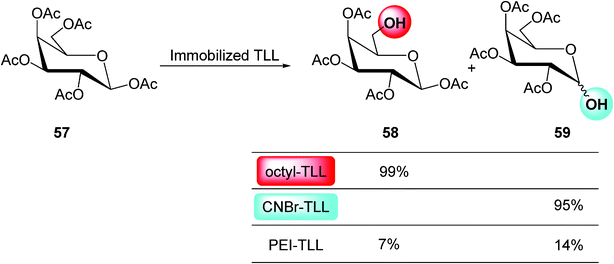 | ||
| Scheme 10 | ||
One biocatalyst (octyl-TLL) was very specific and regioselective toward hydrolysis in C–6 with a >99% yield. However, a second immobilized biocatalyst from the same lipase (CNBr-TLL) was highly specific and regioselective towards the C–1 position. Moreover, a third catalyst (PEI-TLL) was poorly active and not selective at all against one of these positions. Therefore, from this strategy it is possible to obtain very highly selective catalysts (Scheme 10).
This strategy was extended with success to many other lipases demonstrating its potential as a general methodology to efficiently prepare regioselective monodeprotected monosaccharides.50–53
Thus a combinatorial approach—via combination of different pure lipases together with different immobilization protocols—was developed to prepare in one step the regioselective C–6 deprotected tetraacetylated α and β monosaccharides in very high overall yields (Scheme 11).52
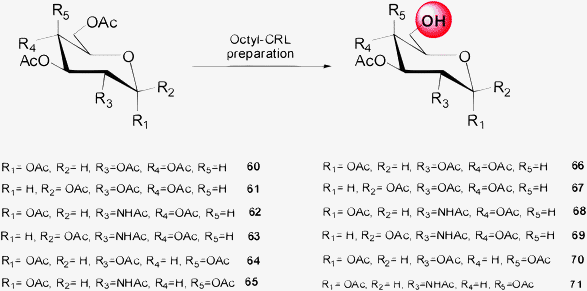 | ||
| Scheme 11 | ||
The per-O-acetylated monosaccharides (60–65) were transformed in the corresponding 6–OH products (66–71) with high overall yields (from 60% to >95%) by selecting the best biocatalyst for each monosaccharide (Scheme 11). In general, the octyl-CRL preparation was the most interesting biocatalyst for regioselective C–6 deprotection of monosaccharides.
A very mild controlled acyl migration applied to the different 6–OH tetraacetylated glycopyranoses (66–71)—by changing from pH 5 (no migration conditions) up to pH 8.5 after removing the immobilized biocatalyst from the reaction—permitted to obtain the subsequent 3–OH and 4–OH products (83–96) in different yields.52 In the glucopyranosidic structure, a high amount of the 4–OH products was preferentially obtained (70–80%) whereas in the galactopyranosidic structure the migration produced an equimolar mix of 3–OH and 4–OH products.
This combinatorial biocatalytic engineering approach was also applied to different glycopyranosides with different substituents in the anomeric position53 and especially to a more reactive carbohydrate derivative such as glycals (Scheme 12).
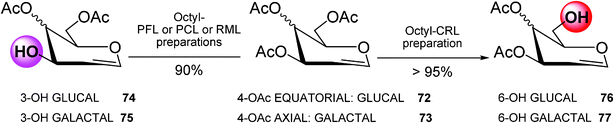 | ||
| Scheme 12 | ||
Thus, by the right selection of the biocatalyst, it was possible to hydrolyze regioselectively the C–6 and C–3 positions of peracetylated glucal (72) and galactal (73) in excellent yields (>95%).
Therefore, the combination of the biocatalytic strategy together with the medium engineering has allowed to prepare efficiently—in “one-pot” step and very mild conditions—a small combinatorial library of different monohydroxy acetylated monosaccharides in high overall yields (Scheme 13).
 | ||
| Scheme 13 | ||
These regioselective deprotected monosaccharides, with a great potential in glycochemistry, were used as a scaffold in the synthesis of several biologically active molecules, such as an lactosamine derivative antitumoral drug55 and tailor-made di- and trisaccharides, direct precursors of Tn epitopes, Mucin glucopeptides or sialyl lewis analogues.56
The synthesis of the antitumoral drug β-O-naphthylmethyl-N-peracetylated lactosamine (104) was recently carried out by a very efficient chemo-enzymatic approach (Scheme 14).55 The 4–OH compound (87), synthesized by the chemo-enzymatic deprotection of peracetylated monosaccharide, was used as the acceptor for the glycosylation reaction with tetra-O-acetyl-α-D-galactopyranosyl trichloroacetimidate (102) as the glycosyl donor, producing the disaccharide (103) in 55% yield after purification. The oxazoline formation and the reaction with 2-napthylmethanol yielded the target product (104) in 20% overall yield (Scheme 14). This synthetic example clearly demonstrates the straightforward potential derived from the application of the chemo-enzymatic approach in glycochemistry. In fact, comparing the entire chemo-enzymatic pathway described above with a “canonical” (obtained through the sole application of orthogonal protecting group strategy) chemical synthesis,56 it is possible to observe an overall yield improvement (20% vs. <5%) together with a noteworthy reduction of the synthetic steps (4 steps vs. 11 steps) (Scheme 14).55
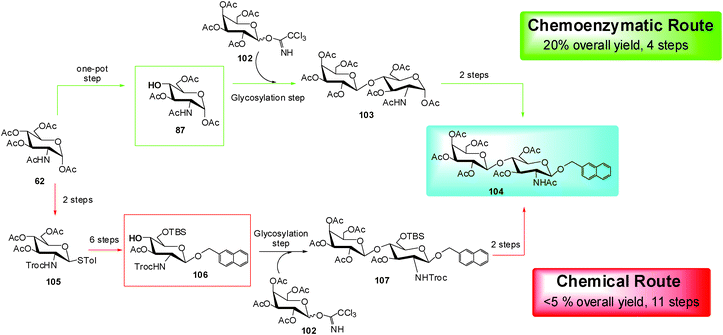 | ||
| Scheme 14 | ||
Tn epitope disaccharides and linear trisaccharides were efficiently synthesized in good yields using some monosaccharides as scaffolds. The novelty of this approach relies on the use of an acetyl group as a unique alcohol protecting group (monoprotective approach).57
The monodeprotected acetylated building-blocks (87) and (68)—obtained by the regioselective chemo-enzymatic approach—were coupled to (102) in the presence of boron trifluoride diethyl etherate (BF3*OEt2) as the activating agent yielding the peracetylated disaccharides (103) and (108) (Scheme 15).
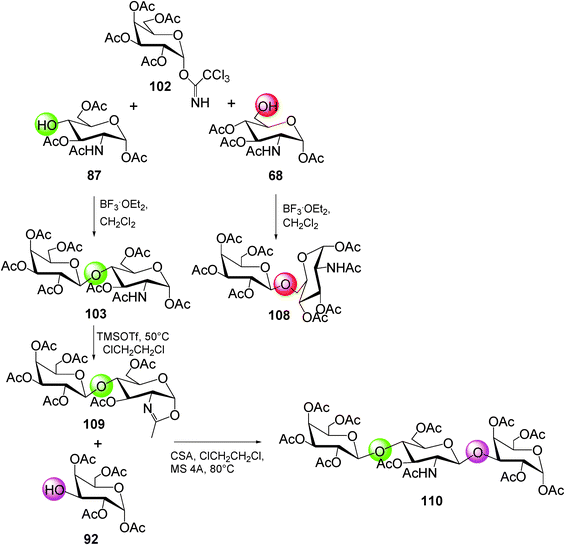 | ||
| Scheme 15 | ||
For the preparation of the linear trisaccharide, the peracetylated lactosamine (103) was converted in the glycosyl donor (109) after the transformation in its oxazoline derivative. Finally, the trisaccharide 110 was synthesized by the regioselective glycosylation using monohydroxy compound 92 as the acceptor (Scheme 15).57
In summary, the great advantage relies on the consideration that starting from a very cheap raw material, sugar, only performing a chemical peracetylation and applying the chemo-enzymatic strategy, it is possible to easily obtain a vast library of high value products useful in glycochemistry.
Recently, a new chemoenzymatic method for the regioselective deprotection of monosaccharide substrates using engineered Bacillus megaterium cytochrome P450 demethylases (P450BM3) has been developed.58 By a fine tuned combination of protein and substrate engineering, Lewis et al.58 obtained a wide range of monodeprotected monosaccharides with different configurations (α or β- gluco, galacto, or mannoside). With this straightforward regioselective and efficient chemoenzymatic procedure it was possible to achieve a set of key valuable intermediates easily convertible in substituted mono- and polysaccharides useful in chemical, biological and medicinal studies. (Scheme 16)
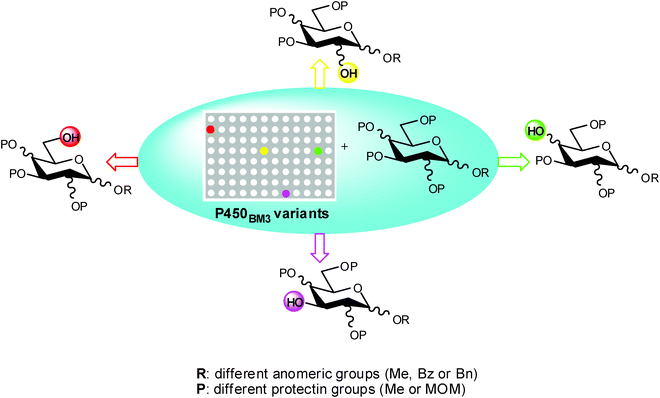 | ||
| Scheme 16 | ||
In fact, the monodemethylated compounds obtained from good to high overall yield (44–98%) have been converted to a variety of biologically relevant products using established chemical transformations (fluorination, to modify the pharmacokinetic properties of molecules or for PET imaging;59 deoxygenation, to obtain deoxy sugars as important therapeutic agents;60 glycosidation, to obtain more complex oligosaccharides) (Scheme 17).
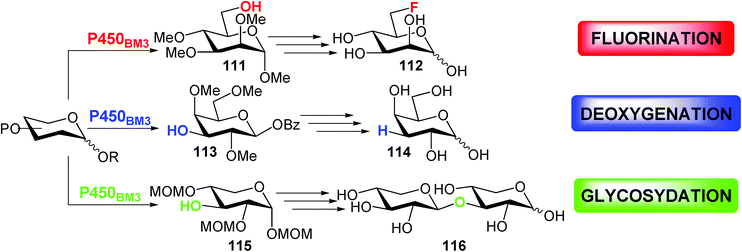 | ||
| Scheme 17 | ||
3. Conclusion
This review has shown novel recent strategies to achieve the synthesis of different monosaccharide derivatives. Using the applicability of known orthogonal protection methods in a different combined way or selective deprotection strategies it has been possible to prepare such a high amount of different activated monosaccharides in excellent yields.The building-blocks prepared with the techniques elucidated above permitted the preparation of a lot of glycoderivatives with important biological effects. For example, among all the examples reported here, the preparation of a glycopeptidic based anticancer vaccine construct,48 the virus binding oligosaccharides involved in the H5N1 avian influenza,27 some glycomimetics with glucosidase inhibitory effects44,55 or some oligosaccharidic cancer epitopes useful in immunolo-stimulation57 have been described.
To sum up, in this work we have reviewed some of the most recent advances that present a great potential in future glycochemistry and offer new straightforward tools for the glycobiology and medicinal chemistry advancement.
Abbreviations
| Et3SIH | triethylsilane |
| TBAF | Tetrabutylammonium fluoride |
| DDQ | 2,3-dichloro-5,6-dicyanobenzoquinone |
| TMS | tetramethylsilane |
| TfO2 | trifluoromethanesulfonyl anhydride |
| TBDPS | tert-butyldiphenylsilyl |
| Fmoc | Fluorenylmethyloxycarbonyl |
| Boc | di-tert-butyl dicarbonate |
| Troc | 2,2,2-trichloroethoxycarbonyl |
| KLH | keyhole limpet hemocyanin |
| MOM | methoxymethyl |
| Bn | benzyl |
| BSP | 1-benzenesulfinyl piperidine |
| Bz | benzoyl |
| CSA | (−) camphor-10-sulphonic acid |
| TBPy | 2,4,6-tri-tert-butylpyrimidine |
| CRL | Candida Rugosa lipase |
| PFL | Pseudomonas fluorescens lipase |
| PCL | Pseudomonas Cepacia lipase |
| RML | Rhizomucor Miehei lipase |
| Octyl | octyl Sepharose |
| PEI | poliethylenimine Sepharose |
| CNBr | cyanogens bromide activated Sepharose |
Acknowledgements
This work has been sponsored by CSIC (Intramural Project 200980I133). The authors are grateful to CSIC for the JAE-DOC contract of Dr Marco Filice. The help and comments from Dr Ángel Berenguer (Instituto de Materiales, Universidad de Alicante) are gratefully recognized.References
- H. E. Murrey and L. C. Hsieh-Wilson, Chem. Rev., 2008, 108, 1708 CrossRef CAS.
- E. Walker-Nasir, A. Kaleem, D. C. Hoessli, A. Khurshid and Nasir ud-Din, Curr. Org. Chem., 2008, 12, 940 CrossRef CAS.
- A. Homann and J. Seibel, Nat. Prod. Rep., 2009, 26, 1555 RSC.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046 CrossRef CAS.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D.M. Haddleton, J. Am. Chem. Soc., 2010, 21, 1940 Search PubMed.
- J. C. López and J. Plumet, Eur. J. Org. Chem., 2011, 1803 CrossRef.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS.
- L. Schofield, M. C. Hewittt, K. Evans, M. A. Slomos and P. H. Seeberger, Nature, 2002, 418, 785 CrossRef CAS.
- C. J. Thibodeaux, C. E. Melançon and H. W. Liu, Nature, 2007, 446, 1008 CrossRef CAS.
- V. Kren and T. Rezanka, FEMS Microbiol. Rev., 2008, 32, 858 CrossRef CAS.
- G. J. Williams, R. W. Gantt and J. S. Thorson, Curr. Opin. Chem. Biol., 2008, 12, 556 CrossRef CAS.
- C. E. Melancon III, W. L. Yu and H. W. Liu, J. Am. Chem. Soc., 2005, 127, 12240 CrossRef.
- D. A. Thayer and C. H. Wong, Chem.–Asian J., 2006, 1, 445 CrossRef CAS.
- H. Y. L. Wang, W. Xin, M. Zhou, T. A. Stueckle, Y. Rojanasakul and G. A. O'Doherty, ACS Med. Chem. Lett., 2011, 2, 73 CrossRef CAS.
- C. T. Campbell and K. J. Yarema, GenomeBiology, 2005, 6, 236 CrossRef.
- A. A. Vyas, H. V. Patel, S. E. Fromholt, M. Heffer-Lauc, K. A. Vyas, J. Dang, M. Schachner and R. L. Schnaar, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8412 CrossRef CAS.
- E. Sidransky, Mol. Genet. Metab., 2004, 83, 6 CrossRef CAS.
- A. Arena, N. Arena, R. Ciurleo, A. de Gregorio, R. Maccari, R. Ottana, B. Pavone, A. Tramice, A. Trincone and M. G. Vigorita, Eur. J. Med. Chem., 2008, 43, 2656 CrossRef CAS.
- A. Nikolakakis, K. Haidara, F. Sauriol, O. Mamer and L. O. Zamir, Bioorg. Med. Chem., 2003, 11, 1551 CrossRef CAS.
- F. S. Ekholm, S. Gyula, W. Janos and R. Leino, Eur. J. Org. Chem., 2011, 6, 104 Search PubMed.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900 CrossRef CAS.
- L. O. Kononov, N. N. Malysheva and A. V. Orlova, Eur. J. Org. Chem., 2009, 611 CrossRef CAS.
- F. Matsumura, N. Oka and T. Wada, Org. Lett., 2008, 10, 5297 CrossRef CAS.
- Y. S. Lu, Q. Li, L. H. Zhang and X. S. Ye, Org. Lett., 2008, 10, 3445 CrossRef CAS.
- J. Y. Baek, Y. J. Joo and K. S. Kim, Tetrahedron Lett., 2008, 49, 4734 CrossRef CAS.
- C. H. Wong, J. Org. Chem., 2005, 70, 4219 CrossRef CAS.
- C. C. Wang, J. C. Lee, S. Y. Luo, S. S. Kulkarni, Y. W. Huang, C. C. Lee, K. L. Chang and S. C. Hung, Nature, 2007, 446, 896 CrossRef CAS.
- (a) P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis 4th ed.John Wiley & Sons, New York, 2007 Search PubMed; (b) C. R. Shie, Z. H. Tzeng, S. S. Kulkarni, B. J. Uang, C. Y. Hsu and S. C. Hung, Angew. Chem., Int. Ed., 2005, 44, 1665 CrossRef CAS.
- Q. Tian, L. Dong, X. Ma, Y. L. Xu, C. Hu, W. Zou and H. W. Shao, J. Org. Chem., 2011, 76, 1045 CrossRef CAS.
- (a) Q. Tian, L. Y. Xu, X. F. Ma, W. Zou and H. W. Shao, Org. Lett., 2010, 12, 540 CrossRef CAS; (b) W. Zou, Z. R. Wang, E. Lacroix, S. H. Wu and H. J. Jennings, Carbohydr. Res., 2001, 334, 223 CrossRef CAS.
- G. L. Thanh, G. Abbenante, G. Adamson, B. Becker, C. Clark, G. Condie, T. Falzun, M. Grathwohl, P. Gupta, M. Hanson, N. Huynh, P. Katavic, K. Kuipers, A. Lam, L. Liu, M. Mann, J. Mason, D. McKeveney, C. Muldoon, A. Pearson, P. Rajaratnam, S. Ryan, G. Tometzki, G. Verquin, J. Waanders, M. West, N. Wilcox, N. Wimmer, A. Yau, J. Zuegg and W. Meutermans, J. Org. Chem., 2010, 75, 197 CrossRef CAS.
- G. L. Thanh, B. Becker, M. Grathwohl, J. Halliday, G. Tometzki, J. Zuegg and W. Meutermans, Drug Discovery Today, 2003, 8, 701 CrossRef.
- P. Dasgupta, Pharmacol. Ther., 2004, 102, 61 CrossRef CAS.
- D. Ferone, M. Boschetti, E. Resmini, M. Giusti, V. Albanese, U. Goglia, M. Albertelli, L. Vera, F. Bianchi and F. Minuto, Ann. N. Y. Acad. Sci., 2006, 1069, 129 CrossRef CAS.
- G. Tulipano and S. Schulz, Eur. J. Endocrinol., 2007, 156, S3 CrossRef CAS.
- D. Cervia and P. Bagnoli, Pharmacol. Ther., 2007, 116, 322 CrossRef CAS.
- J. Guillermet-Guibert, H. Lahlou, P. Cordelier, C. Bousquet, S. Pyronnet and C. Susini, J. Endocrinol. Invest., 2005, 28, 5 CAS.
- P. Barnett, Endocrine, 2003, 20, 255 CrossRef CAS.
- G. Abbenante, B. Becker, S. Blanc, C. Clark, G. Condie, G. Fraser, M. Grathwohl, J. Halliday, S. Henderson, A. Lam, L. Liu, M. Mann, C. Muldoon, A. Pearson, P. Rajaratnam, T. Ramsdale, T. Rossetti, K. Shafer, G. L. Thanh, G. Tometzki, F. Vari, G. Verquin, J. Waanders, M. West, N. Wimmer, A. Yau, J. Zuegg and W. Meutermans, J. Med. Chem., 2010, 53, 5576 CrossRef CAS.
- J. W. Yang, X. P. He, C. Li, L. X. Gao, L. Sheng, J. Xie, X. X. Shi, Y. Tang, J. Li and G. R. Chen, Bioorg. Med. Chem. Lett., 2011, 21, 1092 CrossRef CAS.
- (a) Z. Y. Zhang, Curr. Opin. Chem. Biol., 2001, 5, 416 CrossRef CAS; (b) A. J. Barr, E. Ugochukwu, W. H. Lee, O. N. F. King, P. Filippakopoulos, I. Alfano, P. Savitsky, N. A. Burgess-Brown, S. Müller and S. Knapp, Cell, 2009, 136, 352 CrossRef CAS.
- M. I. García-Moreno, C. Ortiz Mellet and J. M. García-Fernández, Tetrahedron: Asymmetry, 1999, 10, 4271 CrossRef.
- P. Diaz Perez, M. I. García-Moreno, C. Ortiz Mellet and J. M. García-Fernández, Eur. J. Org. Chem., 2005, 2903 CrossRef.
- E. M. Sanchez-Fernandez, R. Rısquez-Cuadro, M. Chasseraud, A. Ahidouch, C. Ortiz Mellet, H. Ouadid-Ahidouch and J. M. Garcıa-Fernandez, Chem. Commun., 2010, 46, 5328 RSC.
- G. Ragupathi, T. K. Park, S. L. Zhang, I. J. Kim, L. Graber, S. Adluri, K. O. Lloyd, S. J. Danishefsky and P. O. Livingston, Angew. Chem., Int. Ed. Engl., 1997, 36, 125 CrossRef CAS.
- S. D. Kuduk, J. B. Schwarz, X. T. Chen, P. W. Glunz, D. Sames, G. Ragupathi, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 1998, 120, 12474 CrossRef CAS.
- G. Ragupathi, F. Koide, P. O. Livingston, Y. S. Cho, E. Atsushi, Q. Wan, M. K. Spassova, S. J. Keding, J. Allen, O. Ouerfelli, R. M. Wilson and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 2715 CrossRef CAS.
- J. Zhu, Q. Wan, G. Ragupathi, C. M. George, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 4151 CrossRef CAS.
- J. Zhu, Q. Wan, D. Lee, G. Yang, M. Spassova, O. Ouerfelli, G. Ragupathi, P. Damani, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 9298 CrossRef CAS.
- J. M. Palomo, M. Filice, R. Fernandez-Lafuente, M. Terreni and J. M. Guisan, Adv. Synth. Catal., 2007, 349, 1969 CrossRef CAS.
- G. Fernandez-Lorente, M. Filice, M. Terreni, J. M. Guisan, R. Fernandez-Lafuente and J. M. Palomo, J. Mol. Catal. B: Enzym., 2008, 51, 110 CrossRef CAS.
- M. Filice, T. Bavaro, R. Fernandez-Lafuente, M. Pregnolato, J. M. Guisan, J. M. Palomo and M. Terreni, Catal. Today, 2009, 140, 11 CrossRef CAS.
- D. S. Rodrigues, A. A. Mendes, M. Filice, R. Fernandez-Lafuente, J. M. Guisan and J. M. Palomo, J. Mol. Catal. B: Enzym., 2009, 58, 36 CrossRef CAS.
- (a) J. M. Palomo, Curr. Org. Synth., 2009, 6, 1 CrossRef CAS; (b) J. M. Palomo, Curr. Bioact. Compd., 2008, 4, 126 CrossRef CAS.
- M. Filice, D. Ubiali, R. Fernandez-Lafuente, G. Fernandez-Lorente, J. M. Guisan, J. M. Palomo and M. Terreni, J. Mol. Catal. B: Enzym., 2008, 52–53, 106 CrossRef CAS.
- T. K. K. Mong, L. V. Lee, J. R. Brown, J. D. Esko and C. H. Wong, ChemBioChem, 2003, 4, 835 CrossRef CAS.
- M. Filice, J. M. Palomo, P. Bonomi, T. Bavaro, R. Fernandez-Lafuente, J. M. Guisan and M. Terreni, Tetrahedron, 2008, 64, 9286 CrossRef CAS.
- J. C. Lewis, S. Bastian, C. S. Bennett, Y. Fu, Y. Mitsuda, M. M. Chen, W. A. Greenberg, C. H. Wong and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16550 CrossRef CAS.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef.
- A. Kirschning, A. F. W. Bechthold and J. Rohr, Top. Curr. Chem., 1997, 188, 1 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |