Transition metal complexes with strong absorption of visible light and long-lived triplet excited states: from molecular design to applications
Jianzhang
Zhao
*,
Shaomin
Ji
,
Wanhua
Wu
,
Wenting
Wu
,
Huimin
Guo
*,
Jifu
Sun
,
Haiyang
Sun
,
Yifan
Liu
,
Qiuting
Li
and
Ling
Huang
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: zhaojzh@dlut.edu.cn; guohm@dlut.edu.cn; Fax: + 86 (0)411 8498 6236; Web: http://finechem.dlut.edu.cn/photochem
First published on 23rd December 2011
Abstract
Transition metal complexes of Ru(II), Pt(II) and Ir(III) with strong absorption of visible light and long-lived T1 excited states were summarized. A design rationale of these complexes, i.e. direct metalation of organic chromophore, was proposed. Alternatively an organic chromophore can be dangled on the peripheral moiety of the coordination center. In both cases the long-lived intraligand triplet excited state (3IL) can be accessed. However, the 3IL excited state is usually emissive for the former case and it is very often non-emissive for the latter case. Two methods used for study of the long-lived triplet excited state, i.e. the time-resolved transient difference absorption spectroscopy and the spin density analysis, are briefly introduced. Preliminary applications of the complexes in luminescent O2 sensing and triplet–triplet annihilation (TTA) upconversions were discussed.
 Jianzhang Zhao | Prof. Jianzhang Zhao received his Ph.D. degree at Jilin University in 2000. Then he carried out postdoctoral research at POSTECH (Pohang, South Korea), Max-Planck Research Unit (Halle, Germany) and University of Bath (U.K.) during 2000–2005. He took up his current position in 2005. His research interests involve fluorescent molecular probes and phosphorescent transition metal complexes, ranging from synthesis to study of photophysical properties with steady-state and time resolved spectroscopy, as well as application of DFT calculations in luminescence studies. |
 Shaomin Ji | Shaomin Ji received her B. S. degree in Applied Chemistry from Hebei University of Technology in 2006. She has been a Ph.D. student in Prof. J. Zhao's group, at Dalian University of Technology during 2006–2011. Her research interests are fluorescent molecular probes and phosphorescent Ru(II) complexes, including synthesis and study of the photophysical properties with steady-state and time-resolved spectroscopy and application of the complexes for luminescent O2 sensing and triplet–triplet annihilation upconversions. |
 Wanhua Wu | Wanhua Wu received her B. S. degree (Applied Chemistry) from Dalian University of Technology in 2008. Since 2008 she has been a Ph.D. student in Prof. J. Zhao's group, at Dalian University of Technology. Her research interests mainly focus on the Pt(II) complexes and organic chromophores that show strong absorption of visible light and long-lived triplet excited states and exploring their applications for O2 sensing and triplet–triplet annihilation based upconversions. |
 Wenting Wu | Wenting Wu received her B. S. degree in Applied Chemistry from Dalian University of Technology in 2007, then she studied at DUT as a Ph.D. student in Prof. J. Zhao's group. Now she is working on the molecular photochemistry & photophysics, especially on the long-lived triplet excited state of Pt(II) complexes and application of the complexes for luminescent O2 sensing and triplet–triplet annihilation upconversions. |
 Huimin Guo | Dr Huimin Guo received her Ph.D. degree in theoretical and physical chemistry under the direction of Prof. Bernhard Dick in 2005 from the University of Regensburg, Germany. She then became a lecturer in the School of Chemistry, Dalian University of Technology, and was promoted to associate professor in 2010. Her research interests mainly focus on fluorescent molecular probes, and photochemical and photophysics of phosphorescent transition metal complexes. |
 Jifu Sun | Jifu Sun received his M. S. degree in Applied Chemistry from China Research Institute of Daily Chemical Industry (Taiyuan) in 2008. Since 2009 he has been a Ph.D. student in Prof. J. Zhao's group, at Dalian University of Technology. Currently he is working on phosphorescent Ir(III) complexes, including synthesis and study of the photophysical properties and application for O2 sensing, upconversion and photo-oxidations. |
1. Introduction
Transition metal complexes have attracted much attention due to their applications in electroluminescence and light-harvesting molecular assemblies.1–2 These complexes, typically Pt(II), Ir(III) and Ru(II), etc., are different from organic fluorophores in that the triplet excited states, instead of the singlet excited states, are populated upon photoexcitation of these complexes.1–8 As a result, long-lived triplet excited states in the microsecond range (μs) were observed for these complexes.1–8In recent years, new applications have been developed for these complexes, such as photovoltaics or photoinduced charge separation,9,10 photocatalysis,11–13 luminescent molecular probes,14,15 or most recently triplet–triplet annihilation (TTA) based upconversions.16–24 It should be pointed out that the requirements for these new applications demand new design rationales for the complexes. For example, (1) intense absorption of visible light is desired for the application of transition metal complexes in photocatalysis, photovoltaics or luminescent molecular probes.15 However, the typical transition metal complexes show weak absorption in the visible region, such as Pt(II) acetylide complexes and Ru(II) complexes, etc.2,25–27 This weak absorption is due to the allowed S0→1MLCT transitions. (2) For new applications of the Ru(II), Pt(II) and Ir(III) complexes in luminescent O2 sensing and TTA upconversions, long-lived T1 excited states are desired for these complexes.20,23,24,28,29 However, the lifetimes of the triplet excited states of the transition metal complexes are usually short (less than 5 μs). Thus, molecular design rationales, with which to access the long-lived triplet excited states, have to be developed. It should be pointed out that for conventional applications of the transition metal complexes, such as electroluminescence, short T1 excited state lifetimes are desired, otherwise the electroluminescence efficiency will be reduced by the saturation effect.30
Concerning these aspects, Ru(II) polyimine complexes with long-lived T1 excited states are relatively more thoroughly investigated than the other complexes.6,31,32 Some photophysical rules for these Ru(II) polyimine complexes may be applicable to complexes such as Pt(II) and Ir(III) complexes. But more room is left for the development of Pt(II) and Ir(III) complexes that show intense visible light absorption and long-lived T1 excited states. Furthermore, the applications of these complexes, such as in luminescent O2 sensing and TTA upconversion are still in early stages although preliminary applications are promising.20,21,24,28,29
Recently, transition metal complexes with intense visible light absorption and a long-lived T1 excited state have been reported. But no design rationales have been unambiguously proposed. Herein we summarize the recent developments in this area. Based on the literature results and our own work, we propose a general rule for design of transition metal complexes that show intense absorption of visible light and long-lived T1 excited states. The contents of this review article cover the cyclometalated Pt(II) and Ir(III) complexes, Pt(II) acetylide complexes and the Ru(II) polyimine complexes. The photophysical and theoretical methods for investigation of the long-lived T1 excited state were also presented.
It should be pointed out that Pt(II)/Pd(II) porphyrin complexes, which show absorption of visible light and long-lived T1 excited states, are not in the scope of this review article, mainly due to the difficulty to tune the absorption and emission wavelength of these complexes. Please refer to recent reviews on the properties and some applications of Pt(II)/Pd(II) porphyrin complexes.19,33–35
2. Ru(II), Pt(II) and Ir(III) complexes that show strong absorption of visible light and long-lived triplet excited states
2.1 Ru(II) polyimine complexes
Ru(II) polyimine complexes are representative transition metal complexes that show visible light absorption and long-lived T1 excited states, with some complexes giving room temperature (RT) phosphorescence.5–7,31,32 However, the absorptions of these complexes usually show only moderate ε values (<20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), and very few phosphorescent Ru(II) complexes show absorption beyond 500 nm (Ru(II) complexes with SCN− ligands are not in the scope of this review. These complexes give strong absorption in the visible range, and have been extensively used for dye-sensitized solar cells, but the photophysics of these complexes are rarely reported). Furthermore, the lifetimes of the T1 excited states of the typical Ru(II) complexes are less than 2 μs.3,6,31,32 Optimization of the coordination geometry of the NˆN ligand can improve the lifetime to some extent, but not significantly.36 Thus new molecular design strategies with consideration of excited states other than the optimization of the coordination geometry of the ligands have to be developed.
000 M−1 cm−1), and very few phosphorescent Ru(II) complexes show absorption beyond 500 nm (Ru(II) complexes with SCN− ligands are not in the scope of this review. These complexes give strong absorption in the visible range, and have been extensively used for dye-sensitized solar cells, but the photophysics of these complexes are rarely reported). Furthermore, the lifetimes of the T1 excited states of the typical Ru(II) complexes are less than 2 μs.3,6,31,32 Optimization of the coordination geometry of the NˆN ligand can improve the lifetime to some extent, but not significantly.36 Thus new molecular design strategies with consideration of excited states other than the optimization of the coordination geometry of the ligands have to be developed.
In the last decade, photophysical investigations on the Ru(II) polyimine complexes that show a long-lived T1 excited state have been extensively carried out.3,6,25,31,32,37–43 In principle, two methods are developed to prolong the lifetimes of triplet excited states of Ru(II) polyimine complexes. The first one is to introduce an organic chromophore into the complex to establish an equilibrium between the intrinsic metal-to-ligand charge transfer (3MLCT) excited state and the ligand-localized 3IL (intraligand or ligand-localized) excited state.25,39–43 In this case the representative example is complex [Ru(pyrenyl-phen)3][PF6]2 (Ru-1, Fig. 1).41 The energy level of the pyrene-localized 3IL state is close to the 3MLCT excited state, thus upon photoexcitation, an equilibrium between the 3IL and 3MLCT can be established. The lifetime of this complex was determined as 148 ± 8 μs (RT).41 Both luminescence and transient absorption methods give the same lifetime. Nanosecond time-resolved transient absorption indicates a 3IL excited state (characteristic triplet absorption of a pyrene moiety, T1→Tn with n > 1). The emissive excited state at RT was recognized as the MLCT state for Ru-1 and the 3IL emissive state was observed at 77 K. This complex shows typical UV-vis absorption of the Ru(II) polyimine complexes, i.e. moderate absorption at 450 nm (ε = ca. 15000 M−1 cm−1. Note that ε for organic chromophores, such as rhodamine, can be as high as 100000 M−1 cm−1).
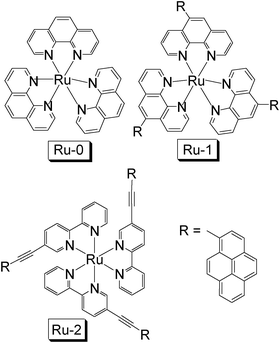 | ||
| Fig. 1 Molecular structures of pyrene-containing complexes Ru-1 and Ru-2, as well as the model complex Ru-0. These compounds are from ref. 37 and 41. | ||
Another strategy to access the long-lived T1 state is to increase the π-conjugation of the NˆN ligand, thus the T1 state was changed from 3MLCT to 3IL state, an example of Ru-2 was presented in Fig. 1.28,37,38,44 Different from Ru-1, the pyrene moieties in Ru-2 were connected to the bpy ligand (bpy = 2,2′-bispyridine) via the ethynyl bond (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C). Thus in Ru-2 the π-conjugation framework of the bpy ligand is extended. The singlet excited state and the triplet excited state of the complex were drastically altered by this ligand modification. Ru-2 shows enhanced absorption in the visible range (ε = 70200 M−1 cm−1 at 420 nm), whereas the model Ru(II) complex without the pyrenyl ethynyl substitution shows an absorption maximum at 450 nm (ε = 12100 M−1 cm−1).
C). Thus in Ru-2 the π-conjugation framework of the bpy ligand is extended. The singlet excited state and the triplet excited state of the complex were drastically altered by this ligand modification. Ru-2 shows enhanced absorption in the visible range (ε = 70200 M−1 cm−1 at 420 nm), whereas the model Ru(II) complex without the pyrenyl ethynyl substitution shows an absorption maximum at 450 nm (ε = 12100 M−1 cm−1).
Furthermore, the RT phosphorescence of Ru-2 is different from the structureless emission band of the Ru(II) polyimine complexes Ru-0 and Ru-1 at ca. 600 nm. The RT phosphorescence of Ru-2 is red-shifted (672 nm) and the emission band is with significant vibrational progression. The most significant difference between Ru-2 and Ru-0 is the lifetime of the RT phosphorescence. The RT phosphorescence lifetime of Ru-2 was determined as 54.3 μs, much longer than the model complex (Ru-0) without the pyrenyl ethynyl substitution (0.84 μs). Note the lifetime of the T1 excited state is prolonged by up to 60-fold. The emission spectra at 77 K, which shows no blue shift compared to the emission at RT (small thermally induced Stokes shift, ΔES), indicates that the emissive excited state is most probably ligand-localized 3IL state, instead of the typical 3MLCT states for the normal Ru(II) polyimine complexes.
Nanosecond time-resolved transient difference absorption of the complex Ru-2 is drastically different from that of the model complex without the pyrenyl ethynyl substitution, thus the triplet excited state of Ru-2 upon photoexcitation is recognized as the pyrene ethynyl-localized 3IL excited state. The prolonged lifetime of the 3IL excited state compared to the relatively short lived 3MLCT excited state of the Ru(II) polyimine complexes can be attributed to the less involvement of the Ru(II) in the 3IL state, thus the heavy atom effect of Ru(II) in 3IL state is less than that for 3MLCT, as a result, the ISC is reduced and the decay rate constants of the T1→S0 transition is decreased.
The effect of ligand modification on the photophysical properties can be illustrated by Ru-3, Ru-4 and Ru-5 (Fig. 2 and Fig. 3).28 The connection of the pyrene moiety to the phenanthroline ligand through a C–C single bond induces small changes to the UV-vis absorption and the emission. With an ethynyl bond linker between pyrene and phenanthroline, however, the absorption in the 400–500 nm range was enhanced compared to that of the model complex Ru-3. Furthermore, the emission wavelength of Ru-4 is similar to that of the model complex Ru-3, whereas the emission of Ru-5 is much more red-shifted and structured compared to that of Ru-3, indicating an 3IL emissive excited state for Ru-5. The emissive triplet excited state of Ru-4 can be assigned to 3MLCT, which is in equilibrium with the 3IL state. Both Ru-4 (τ = 9.22 μs) and Ru-5 (τ = 58.4 μs) show much longer T1 excited state lifetimes than that of Ru-3 (τ = 0.45 μs).28
 | ||
| Fig. 2 Pyrene ethynyl containing Ru(II) complex Ru-5, pyrene-containing Ru-4 and the model complex Ru-3. These compounds are from ref. 28. | ||
 | ||
| Fig. 3 UV-vis absorption and emission spectra of Ru(II) polypyridine complexes Ru-3, Ru-4 and Ru-5. (a) UV-vis absorption spectra, (b) Normalized emission spectral of Ru-3, λex = 450 nm; Ru-4, λex = 450 nm; Ru-5, λex = 418 nm. In CH3CN, 1.0 × 10−5 M. 25 °C. Reproduced from ref. 28 with permission . | ||
The photophysical processes involved in the both kinds of Ru(II) polyimine complexes that show long-lived T1 excited states can be summarized in Scheme 1. For Ru-1 and Ru-4, the establishment of the 3IL↔3MLCT state equilibrium prolonged the lifetime of the 3MLCT excited state, whereas the long-lived T1 excited states of Ru-2 and Ru-5 are attributed to the transition from the normal 3MLCT state to the ligand-localized 3IL state. Note that the T1 energy level of the ligand in Ru-2 and Ru-5 must be appropriately lower than the 3MLCT state. In some cases the equilibrium between the two states was confirmed by the temperature-dependent emission of the complex.44 The energy level of the organic chromophore is crucial for the establishment of the equilibrium. The significantly lower energy level of the ligand will lead to population of the 3IL excited state.
 | ||
| Scheme 1 Simplified energy level diagram and the emission states for Ru polypyridine dyad complexes. (a) Normal 3MLCT emission (Ru-3); (b) 3MLCT emission with triplet states equilibrium (Ru-4) and (c) 3IL/3LLCT emission (Ru-5). Reproduced with permission from ref. 28. | ||
These two approaches to access the long-lived triplet excited states have been widely used for the preparation of Ru(II) polyimine complexes.3,6,31,32,37–43,45,46 However, it should be pointed out that the photophysics of the Ru(II) organic chromophore dyads are elusive, that is, the introduction of an organic chromophore does not necessarily guarantee effective enhanced absorption (namely, the harvested excitation energy can be efficiently funnelled to the T1 state) or prolonged T1 excited states.
Borondipyrromethene (BODIPY) is an interesting fluorophore due to its strong absorption in the visible range, high fluorescence quantum yield and good photostability.47,48 the BODIPY moiety has been incorporated into Ru(II) polyimine complexes, probably with the intention to access the Ru(II) complexes with intense absorption of visible light and long-lived triplet excited state. However, very often the Ru(II)-BODIPY dyads demonstrated different photophysical properties from those of the Ru(II)-pyrene dyads. For example, Ru-6 and Ru-7 (Fig. 4) displayed no RT luminescence, that is, neither the fluorescence of the BODIPY ligand, nor the phosphorescence of 3MLCT or 3IL was observed at RT.49 At 77 K (in a rigid matrix), fluorescence of the BODIPY ligand at 540 nm and phosphorescence of BODIPY at 774 nm were observed. It should be pointed out that Ru-6 and Ru-7 show intense absorption at ca. 520 nm (ε > 60000 M−1 cm−1). Considering the connection profile of the BODIPY chromophore to the NˆN ligand, we propose that the π-conjugation frameworks of the chromophore BODIPY and the Ru(II) coordination center are isolated from each other. Since the T1 state energy level of the BODIPY is significantly lower than that of Ru(II) coordination center, thus quenching of the 3MLCT emission of the typical Ru(II) complexes in Ru-6 and Ru-7 can be rationalized by the 3MLCT→3IL internal conversion (IC). The distance between the Ru(II) center and the BODIPY fluorophore is large and the π-conjugation is disrupted by the phenyl ring, thus we anticipated that the 1IL→3IL transition is noneffective. This is demonstrated by the observation of the fluorescence of BODIPY ligand at 77 K. The ISC efficiency was not reported for Ru-6 and Ru-7.49 Thus, the intense absorption does not necessarily always produce the triplet excited state of the Ru(II) complexes efficiently.
 | ||
| Fig. 4 Ru(II) polyimine complexes containing BODIPY chromophore, Ru-6 and Ru-7. These compounds are from ref. 49. | ||
We propose that the photophysical methods for accessing the Ru(II) complexes that show intense absorption of visible light and long-lived triplet excited states can be extended to other transition metal complexes, such as Pt(II) complexes and Ir(III) complexes, etc. Currently the development of Pt(II)/Ir(III) complexes that show intense visible light absorption and long-lived triplet excited states are still in its early age.
2.2 NˆNPt(II) acetylide complexes
Pt(II) acetylide complexes are usually phosphorescent at RT and the fluorescence of the ligands are completely quenched in the complexes, indicating the efficient ISC process.2,25,26,45,50 The principle photophysical process of the Pt(II) acetylide complexes are similar to that of the Ru(II) polyimine complexes, that is, the excitation into the 1MLCT excited state is followed by an efficient ISC to the triplet excited state, which was identified as a 3MLCT/3LLCT transition. Two prominent photophysical features should be noted for Pt(II) acetylide complexes, i.e. the high phosphorescence quantum yield (up to 40%) and the readily tunable photophysical properties by simply changing the structure of the acetylide ligand.2,25,26 However, the disadvantages of the normal NˆNPt(II) bisacetylide complexes are their poor absorption in the visible region (Pt-1, ε = ca. 2500 M−1 cm−1 at 450 nm. Fig. 5) and the short-lived T1 excited state, usually less than 5.0 μs. | ||
| Fig. 5 The dbbpy Pt(II) bisphenyl acetylide complex Pt-1 and pyrenyl complex Pt-2. These complexes are from ref. 51. | ||
In 2003, Castellano et al. reported a dbbpy Pt(II) bisacetylide with 1-ethynylpyrene ligand (Pt-2, where dbbpy = 4,4′-di(tert-butyl)-2,2′-bipyridine. Fig. 5).51 In this connection strategy, the π-core of the pyrene unit is directly coupled to the Pt(II) center, thus the heavy atom effect of Pt atom can be maximized. The UV-vis absorption was enhanced in the visible range. For example, the ε value at 450 nm is ca. 10300 M−1 cm−1. Notably, the RT emission of Pt-2 (λem = 660 nm) is drastically different from that of Pt-1. The emission band of Pt-2 is red-shifted by 100 nm compared to Pt-1. Furthermore, the emission band of Pt-2 is more structured, that is, with significant vibrational progression. For Pt-1, however, the characteristic structureless emission band was observed at 570 nm. The phosphorescence quantum yield of Pt-2 is 1.1%, which is much lower than that of Pt-1 (11%).25 The lifetime of the triplet excited state of Pt-2 was determined as 48.5 μs, which is much longer than that of Pt-1 (1.25 μs).
The emissive triplet excited state of Pt-2 was assigned to a pyrenyl-localized 3IL excited state, supported by the comparison of the emission spectra at 77 K and that at RT (very small thermally induced Stokes shift, ΔES, of 70 cm−1 was observed). Under the same conditions, a large ΔES value of 2800 cm−1 was observed for Pt-1. Nanosecond time-resolved transient difference absorption spectra also support the assignment of the 3IL excited state for Pt-2, for which the characteristic triplet absorption of pyrene moiety (T1→Tn) in the range of 410—700 nm was found, which is absent in the transient absorption of Pt-1. The population of the emissive 3IL excited state of Pt-2 can be attributed to the much lower 3IL state energy level of the ligand than the normal 3MLCT state of the Pt(II) acetylide coordination centre (approximated as 550 nm).
In 2008, Jean-Luc Fillaut et al. reported the flavone based Pt(II) acetylide complexes Pt-3, Pt-4 and Pt-5 (Fig. 6).52 The absorptions of these complexes were enhanced compared to that of the model complexes, such as Pt-1 (Fig. 5). The emissions of these complexes were observed as structureless bands at ca. 600 nm. The phosphorescence lifetimes were 12–21 μs.
 | ||
| Fig. 6 Flavone-based alkyne ligands and the related Pt(II) complexes Pt-3, Pt-4 and Pt-5. These compounds are from ref. 52. | ||
In 2008, Ziessel and Castellano reported the Pt(II) peryleneacetylide complexes Pt-6–Pt-10 (Fig. 7).53 With the peryleneacetylide ligand, the UV-vis absorption of the Pt(II) complexes are greatly enhanced compared to Pt-1 and Pt-6. For example, the absorption of the Pt(II) peryleneacetylide complexes generally give ε > 40000 M−1 cm−1 in the range of 400–500 nm. No phosphorescence was observed for these peryleneacetylide Pt(II) complexes. Nanosecond time-resolved transient difference absorption demonstrated that the lowest-lying triplet excited states of the complexes are the peryleneacetylide localized 3IL excited state. This assignment was further proved with the complexes containing phosphine ligands (Pt-8 and Pt-10), for which population of the MLCT state is impossible. This is demonstrated by the superimposable transient absorption spectra of Pt-7 and Pt-8. For Pt-8, no MLCT state exists thus the observed long-lived transient was exclusively assigned to the perylene localized 3IL excited state. T1 excited state lifetimes of 3.0 μs, 12.6 μs, 5.7 μs and 23.6 μs were observed for the complexes Pt-7–Pt-10, compared to the lifetime of the T1 excited state of the model complexes Pt-1 (1.2 μs) and Pt-6 (3.1 μs). However, no conclusive explanation was proposed for the origin of the different lifetimes of these complexes, though the acetylide ligand is the same.53 Herein the elusive photophysics of the Pt(II) acetylide complexes were demonstrated by the lack of phosphorescence of the complexes where the perylene moiety is directly metalated, which is similar to Pt-2 (Fig. 5). Pt-2 is phosphorescent at RT.51
 | ||
| Fig. 7 Pt(II) peryleneacetylide complexes Pt-7, Pt-8, Pt-9, Pt-10 and the model complex Pt-6. These compounds are from ref. 53. | ||
BODIPY is well-known for its intense absorption in the visible region (the unsubstituted BODIPY chromophore absorbs at ca. 500 nm, with ε up to 80000 M−1 cm−1). Campagna and Ziessel reported a NˆNˆNPt(II) acetylide complex with the BODIPY acetylide (Pt-11), and the photophysical properties are compared with the model complex Pt-12 (Fig. 8).54 Compared to the model complex Pt-12, which gives weak absorption in the visible region (ε = 1850 M−1 cm−1 at 443 nm), complex Pt-11 gives strong absorption at 523 nm (ε = 54610 M−1 cm−1) while Pt-12 gives no absorption at 523 nm at all. However, intense absorption does not guarantee luminescence or long-lived T1 excited states. Model complex Pt-12 gives 3MLCT emission at 635 nm at RT, with a RT lifetime of 310 ns. For the complex Pt-11, however, the fluorescence of the BODIPY ligand was observed at 536 nm, which is supported by the short luminescence lifetime of 3.3 ns. Neither significant 3MLCT emission nor the 3IL emission of BODIPY was observed at RT for Pt-11. The quenched 3MLCT emission in Pt-11 is attributed to the energy transfer from 3MLCT to the BODIPY localized 3IL excited state. At 77 K, fluorescence of the BODIPY ligand at 535 nm (τ = 6.0 ns) and the phosphorescence of BODIPY at 777 nm (τ = 74 ms) were observed. No time-resolved transient difference absorption was carried out for the complexes.54 We propose that the lack of RT phosphorescence of the BODIPY ligand is due to the weak ISC effect because the π-core of the BODIPY moiety is not directly attached to the Pt(II) centre. The phenyl ring in Pt-11 isolates the Pt(II) coordination centre and the BODIPY π-conjugation core.
 | ||
| Fig. 8 BODIPY acetylide-containing NˆNˆNPt(II) acetylide complex Pt-11 and the model complex Pt-12. These compounds are from ref. 54. | ||
Perylenediimide (PDI) is a versatile chromophore for its strong absorption in the visible region and the fluorescence property. PDI was also attached to the Pt(II) centre via the acetylide linkers (Pt-13 and Pt-14. Fig. 9).55 Complexes Pt-13 and Pt-14 show intense absorption at 570 and 573 nm (ε is ca. 65500–68000 M−1 cm−1).55 However, no emissions were observed for these complexes. Furthermore, T1 excited state lifetimes, measured by the nanosecond time-resolved transient difference absorption spectroscopy, are 0.25 μs and 1.0 μs for Pt-13 and Pt-14, respectively. Thus attaching organic chromophores does not necessarily produce long-lived T1 excited states.
 | ||
| Fig. 9 Pt(II) complexes with bisperylenediimide (PDI) acetylide ligands (Pt-13 and Pt-14). These compounds are from ref. 55. | ||
Inspired by the design rationale of Pt-13, recently we prepared a naphthalenediimide (NDI) acetylide based Pt(II) complex (Pt-15, Fig. 10).23aPt-15 shows intense absorption at 583 nm (ε = 31300 M−1 cm−1). The solution of the complex shows a purple color compared to the light yellow color of the model complex solution. The complex is phosphorescent at RT (emissive wavelength is 784 nm). An emission band at a shorter wavelength was observed at 603 nm, which is tentatively assigned to the fluorescence of the ligand.56 The singlet and triplet feature of the two emission bands are assigned by their sensitivity toward O2. Phosphorescence can be significantly quenched by O2, whereas the fluorescence is inert to O2 in most cases. We proved that the T1 excited state of Pt-15 is NDI localized 3IL excited state by using the nanosecond time-resolved transient difference absorption, in which a significant bleaching of the NDI absorption at 580 nm was observed. A RT lifetime of 22.3 μs was observed for Pt-15 with the transient absorption spectra, which is much longer than that of the model complex Pt-1 (1.2 μs).
 | ||
| Fig. 10 Pt(II) complexes with Naphthalenediimide (NDI) acetylide (Pt-15). The spin density distributions of Pt-15 and the model complex Pt-1 are also presented. DFT calculation was performed at B3LYP/6-31G/LANL2DZ level with Gaussian 09W. Please note that the alkyl chains of Pt-NDI were simplified as methyl groups to reduce the computation time. Adapt from ref. 23a with permission. | ||
The assignment of the T1 excited state of Pt-15 as the 3IL excited state was corroborated by DFT calculations on the spin density surface of the complexes (Fig. 10). For the model complex Pt-1, it is known that the T1 excited state is a 3MLCT/3LLCT mixed feature.25–27 The spin density is distributed on phenylacetylide ligands, the Pt(II) center and dbbpy ligand, thus the spin density analysis indicated a 3MLCT/3LLCT feature for the T1. For Pt-15, the spin density is mainly distributed on the NDI ligand, the Pt(II) atom contributes very little to the spin density and the dbbpy ligand makes almost no contribution (Fig. 10). Thus the 3IL T1 excited state can be assigned for Pt-15.23a Therefore, analysis of the spin density will be useful for the design of transition metal complexes to access the long-lived 3IL excited state of organic chromophores.
Naphthalimide (NI) is a robust fluorophore and has been extensively used in fluorescent molecular probes.57,58 However, the application of a NI moiety in transition metal complexes or phosphorescence is rare.59,60 Previously a NI-containing Ru(II) complex was prepared, which shows a long-lived 3MLCT state, due to the equilibrium between the 3MLCT state and the 3IL state.59 No NI localized emission was observed. Recently we prepared the complex Pt-16, with the NI moiety directly attached to the Pt(II) centre via an acetylide linker, to maximize the heavy atom effect of Pt(II).61 The UV-vis absorption of the complex is greatly enhanced in the visible region (ε = 60000 M−1 cm−1 at 424 nm). A structured emission band at 616 nm was observed, which is different from the structureless emission band of the model complex Pt-1 at 570 nm. With nanosecond time-resolved transient difference absorption spectroscopy, the T1 excited state was assigned as the 3IL excited state, which is localized on the NI moiety, because bleaching of the NI absorption band was observed. This assignment is supported by the emission spectrum at 77 K, which shows a very small thermally induced Stokes shift of 131 cm−1. By comparison, the model complex Pt-1, with the 3MLCT excited state, shows a large thermally induced Stokes shift (ΔES) of 2470 cm−1 under the same experimental conditions. The 3IL excited state is also supported by the spin density analysis (Fig. 11). The spin density is mainly localized on the NI acetylide ligand with minor contribution from the Pt(II) centre. This is in agreement with the 3IL assignment. The RT lifetime of the complex Pt-16 was determined as 124 μs, compared to the 1.2 μs for the model complex Pt-1.
 | ||
| Fig. 11 Pt(II) complex with naphthalimide (NI) acetylide ligands (Pt-16) and the spin density surface of the complex. DFT calculation was performed at B3LYP/6-31G/LANL2DZ level with Gaussian 09W. Please note that the alkyl chains of NI moiety were simplified as methyl groups to reduce the computation time. This compound is from ref. 61. | ||
Recently we prepared a dbbpy Pt(II) bisacetylide complex (Pt-17) with the coumarin acetylide ligand (Fig. 12).22Pt-17 shows enhanced absorption in the visible region (ε = 32300 M−1 cm−1 at 414 nm). A structured emission band at 623 nm was observed, which is red shifted by 57 nm compared to the emission of the model complex Pt-1. The emission spectrum at 77 K was measured and structured emission bands with small thermally induced Stokes shift of 182 cm−1 were observed for Pt-17. Thus a T1 state with 3IL as the major components was proposed. Complex Pt-17 shows a lifetime of 2.52 μs, which is not significantly longer than that of the model complex Pt-1 (1.27 μs with the same experimental conditions). The spin density surface of Pt-17 was also investigated (Fig. 12). Different from that of Pt-15 or Pt-16, the Pt(II) centre and the dbbpy ligand contribute significantly to the spin density surface of Pt-17. Thus we propose that the 3IL excited state is mixed with significant MLCT feature, which may be responsible for the short lifetime of the T1 state of Pt-17 to some extent.22
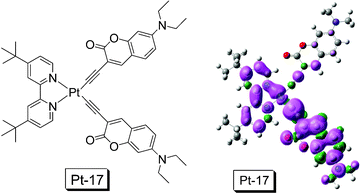 | ||
| Fig. 12 Pt(II) complex Pt-17 with the coumarin acetylide ligands and the spin density surface of the complex. DFT calculation was performed at B3LYP/6-31G/LANL2DZ level with Gaussian 09W. This compound is from ref. 22. | ||
Since it is synthetically feasible to prepare NˆNPt(II) bisacetylide complexes, acetylide ligands can be used to prepare this kind of complexes, and with the large amount of organic chromophores that can be easily converted to acetylides, we believe that a lot of Pt(II) bisacetylide complexes can be prepared with this approach.
Recently we reported a dbbpy Pt(II) bisacetylide complex with rhodamine moiety as the light-harvesting antenna, in order to enhance the absorption and to access the long-lived 3IL excited state (rhodamine-localized) (Pt-18. Fig. 13).23b The complex shows strong absorption at 556 nm (ε = 185800 M−1 cm−1). The difference between the absorption of the complex and the model complex Pt-1 is substantial.23b By comparison, the intense absorption of Pt-18 at 556 nm is due to the rhodamine acetylide ligand. Interestingly, only fluorescence (580 nm) was observed for Pt-18 and no phosphorescence was observed for Pt-18 either at RT or 77 K. The assignment of fluorescence is based on the small Stokes shift of the emission band (24 nm), short luminescence lifetime (2.50 ns) and its insensitivity to O2.23b
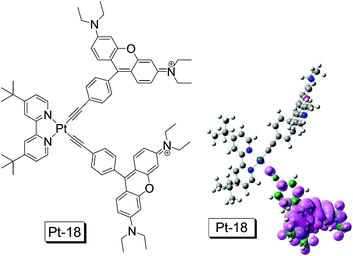 | ||
| Fig. 13
NˆNPt(II) acetylide complex (Pt-18) containing rhodamine moiety. The complex shows strong absorption at 556 nm (ε = 185800 M−1 cm−1) and long-lived non-emissive 3IL excited state was observed (τT = 83.0 μs). DFT calculation was performed at B3LYP/6-31G/LANL2DZ level with Gaussian 09W. The complex is from ref. 23b. | ||
Interestingly, nanosecond time-resolved transient difference absorption spectra show that the rhodamine-localized triplet excited state was populated upon excitation of Pt-18. The lifetime of the triplet excited state is 83.0 μs. This assignment of the triplet excited state as 3IL state was supported by the position of the bleaching band and DFT calculations (spin density analysis of the triplet state of the complex, Fig. 13).
Pt-18 was used as triplet sensitizer for TTA upconversion with perylene as the triplet acceptor and an upconversion quantum yield of 11.2% was observed. Note the overall upconversion capability of Pt-18 is significant, due to its strong absorption at 556 nm (ε = 185800 M−1 cm−1).
Although BODIPY has been used in Ru(II) or Pt(II) complexes, the RT phosphorescence of BODIPY moiety was never observed. Even worse, very often the complexes were non-luminescent.49,54 After examination of the molecular structures, we realized that the π-conjugation core of the BODIPY chromophores were not directly metalated, or connected to the metal coordination center. In order to address this challenge, we recently designed complex Pt-19.23c The BODIPY core in Pt-19 is directly connected to the Pt(II) coordination center viaCC conjugation bonds, which is different from the complexes with the connection at the phenyl moiety (Pt-11, Fig. 8).49,54 We propose the heavy-atom effect of Pt(II) on the BODIPY core can be maximized with the structural profile of Pt-19, than that in Pt-11 (Fig. 8). Pt-19 shows a strong absorption of visible light (ε = 53800 M−1 cm−1 at 574 nm). Room temperature phosphorescence of the BODIPY ligand at 770 nm was observed. Another emission band at 660 nm was also observed. The phosphorescence quantum yield is 3.5% (RT).23c To the best of our knowledge, this is the first report of the RT phosphorescence of BODIPY chromophore. In comparison, the model complex Pt-20 shows the normal photophysical properties. Pt-19 was used as triplet sensitizer for TTA upconversion.
Nanosecond time-resolved transient difference absorption spectroscopy and the spin density surfaces of Pt-19 and Pt-20 were studied (Fig. 14). The results indicated that the T1 state of Pt-19 is localized on BODIPY ligand, i.e., an 3IL excited state. Notably the lifetime of the 3IL state is 128.4 μs (RT). In comparison the T1 state lifetime of Pt-20 is 6.5 μs.
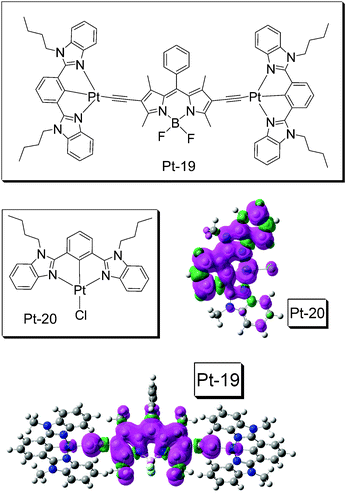 | ||
| Fig. 14
NˆCˆNPt(II) acetylide complex (Pt-19) containing a BODIPY moiety. The complex shows strong absorption at 574 nm (ε = 53800 M−1 cm−1) and a long-lived emissive 3IL excited state was observed (τT = 128.4 μs, Φp = 3.5%). The model complex Pt-20 is also presented (absorption maximum is at 414 nm with ε = 16200 M−1 cm−1, τT = 6.5 μs). DFT calculations were performed at B3LYP/6-31G/LANL2DZ level with Gaussian 09W. Adapted from ref. 23c with permission. | ||
These results confirmed that the connection of an organic chromophore to a metal coordination center is a straight forward method to prepare transition metal complexes that show strong absorption of visible light and long-lived T1 excited states.
2.3 CˆN cyclometalated Pt(II) complexes
Cyclometalated Pt(II) complexes, typically with the structural profile of CˆNPt(II) acac (where CˆN is the cyclometalation ligand, such as 2-phenylpyridine, ppy, and acac = acetyl acetonate), are another kind of representative phosphorescent Pt(II) complexes.2,62–64 Similar to the Pt(II) acetylide complexes, cyclometalated Pt(II) complexes usually show weak absorption of visible light, and the lifetime of the triplet excited state is less than 5 μs. Thus it is desirable to prepare cyclometalated Pt(II) complexes that show intense absorption of visible light and long-lived triplet excited state.The model complex ppy Pt(II) (acac) (where ppy = 2-phenylpyridine, Pt-21) gives weak absorption in the visible range (ε = 18000 M−1 cm−1 at 430 nm) (Fig. 15). A structured emission band at 486 nm was found. It was proposed that the T1 state of this complex is a 3MLCT/3IL mixed feature. The lifetime of the complex is 2.6 μs (RT).65 With the 2-phenylbenzothiozole ligand (Pt-22), the UV-vis absorption in the visible region was improved (ε = 30000 M−1 cm−1 at 443 nm). But the lifetime (0.34 μs) is shorter than that of Pt-21.21 With the coumarin ligand directly cyclometalated, that is, the coumarin moiety as the C donor of the cyclometalated CˆPtˆN bond, the absorption in the visible region of Pt-23 is significantly improved (ε = 51000 M−1 cm−1 at 496 nm) compared to Pt-21. Structured emission bands at 589 nm and 650 nm were observed, indicating that the 3IL feature is significant for the T1 excited state of Pt-23. Notably, the phosphorescence lifetime of this complex is 27.9 μs, which is significantly longer than the model complex Pt-21 (2.6 μs). Furthermore, complex Pt-23 shows a high phosphorescence quantum yield of 25%, higher than that of the model complex Pt-21 (Φ = 15%).
 | ||
| Fig. 15 The model cyclometalated CˆNPt(II) acac complex Pt-21, complex with 2-phenylbenzothiozolato ligand (Pt-22) and with 3-(2-benzothiazolyl)-7-(diethylamino) coumarin ligand (Pt-23). These compounds are from ref. 21, 65. | ||
In order to probe the relationship between the ligand structure and the property of the complexes, we prepared two coumarin-containing complexes Pt-24 and Pt-25, which are analogues of Pt-23 (Fig. 16).21Pt-24 has a similar π-conjugation to that of Pt-23, but without the 7-diethylamino group on the coumarin moiety. Pt-25 has a larger π-conjugation in the CˆN ligand than that of Pt-21. We found that the coumarin ligands in Pt-23, Pt-24 and Pt-25 show absorption bands with similar intensity, but the ligands of Pt-24 and Pt-25 show blue-shifted absorption compared to that of Pt-23 (Fig. 17a).
 | ||
| Fig. 16 Cyclometalated Pt(II) complexes with 3-(2-benzothiazolyl)-coumarin ligand (Pt-24), and 3-(2-benzothiazolyl)-6,7-Benzocoumarin ligand (Pt-25). The complexes are from ref. 21. | ||
 | ||
| Fig. 17 UV-vis absorption spectra of (a) the CˆN ligands L-22, L-23, L-24 and L-25 (corresponding to the complexes structures in Fig. 15 and Fig. 16) and (b) the complexes Pt-22, Pt-23, Pt-24 and Pt-25. In CH2Cl2 (c = 1.0×10−5 M; 25 °C). Reproduced from ref. 21 with permission. | ||
After cyclometalation with Pt(II), the intense absorption of the ligand of Pt-23 persists, and the absorption maxima are red-shifted compared to the free ligands (Fig. 17b). For Pt-24 and Pt-25, however, the absorption intensity is decreased compared to that of the free ligands (Fig. 17b). Furthermore, we found that the lifetimes of complexes Pt-24 (τ = 1.59 μs) and Pt-25 (τ = 1.28 μs) are much shorter than that of Pt-23.21 Thus the subtle change of the ligand will have a significant effect on the photophysical properties of the cyclometalation Pt(II) complexes. These results also indicate that the ideal ligands are those which show intense absorption of visible light.
Yip et al. reported the pyrene-containing Pt(II) complexes (Fig. 18).66 Among the complexes, Pt-27, the complex with two C–Pt bonds, shows the most red-shifted absorption (ε = 52100 M−1 cm−1 at 400 nm). The effect of the metalation on the emission is remarkable (Fig. 19). For example, the phosphorescence is much more significant with formation of the C–Pt bonds, Pt-26 and Pt-27, with one and two C–Pt bonds, respectively, show significant phosphorescence at 611− 627 nm. The residue fluorescence of the ligand below 450 nm is weak. For Pt-28 and Pt-29, however, the phosphorescence beyond 600 nm is weak and the fluorescence at 400 nm is significant (Fig. 19b). Pt-26 and Pt-27 show a long-lived emissive T1 excited state of 31.3 μs and 63.7 μs, respectively.
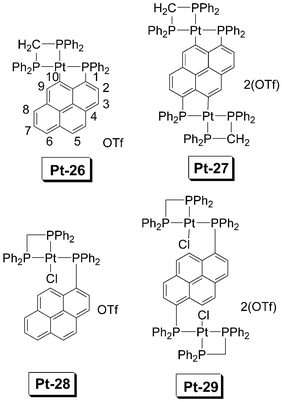 | ||
| Fig. 18 Pyrene-containing Pt(II) complexes Pt-26, Pt-27, Pt-28 and Pt-29. The compounds are from ref. 66. | ||
 | ||
| Fig. 19 (a) Emission spectra of deaerated MeCN solutions of Pt-26 (red) and Pt-27 (black) at room temperature. Excitation wavelength: 320 nm. (b) Emission spectra of Pt-28 (red) and Pt-29 (black) in deaerated CH3CN solution at room temperature. Excitation wavelength: 320 nm. Reproduced from ref. 66 with permission. | ||
Recently we prepared pyrene-containing complexes Pt-30, Pt-31 and Pt-32 (Fig. 20).67Pt-30, the complex with direct cyclometalation of the pyrene moiety, gives the most significant absorption (ε = 34300 M−1 cm−1 at 415 nm). Different from the previous report,66 our results show that all the complexes Pt-30, Pt-31 and Pt-32 give phosphorescence in the range 600 nm—800 nm. The fluorescence due to the ligands are weak for these complexes. This is especially interesting for Pt-31, in which the pyrene unit is not directly metalated. Pt-31 and Pt-32 show long-lived emissive triplet excited state with lifetimes of 6.22 μs and 15.8 μs, respectively. For the model complex Pt-33, a lifetime of the emissive T1 excited state of 5.6 μs was observed.
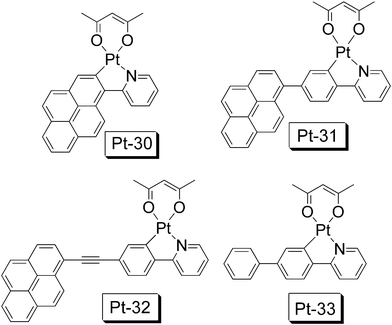 | ||
| Fig. 20 CˆN cyclometalated Pt(II) complexes with a pyrene moiety (Pt-30, Pt-31) or pyrenyl ethynyl ligand (Pt-32). The model complex Pt-33 is also presented. The compounds are from ref. 67. | ||
Recently we prepared naphthalimide (NI) and naphthalene-containing complexes Pt-34, Pt-35, Pt-36 and Pt-37 (Fig. 21).29 The design rationales for the complexes are to access the long-lived 3IL excited state by extension of the π-conjugation of the CˆN ligands. The complexes with the keto-ligands, i.e.Pt-35 and Pt-37, were obtained as the side-product of the metalation of the ethynylene ligands with K2PtCl4. It was found the complexes Pt-34 shows the most intense absorption (ε = 33700 M−1 cm−1 at 390 nm). Its analogue Pt-35 shows a much weaker absorption (ε = 4120 M−1 cm−1 at 400 nm). Complex Pt-34 gives RT phosphorescence at 638 nm, whereas other complexes show blue-shifted RT phosphorescence in the range 530–600 nm. The phosphorescence lifetimes of the complexes were determined as 6.6 μs, 25.5 μs, 15.8 μs and 0.86 μs, respectively. In this case there is no clear trend in the lifetimes of the complexes.
 | ||
| Fig. 21 Cyclometalated Pt(II) acac complex with naphthalimide (NI) (Pt-34 and Pt-35) and naphthalene ligand (Pt-36 and Pt-37). The compounds are from ref. 29. | ||
Luisa De Cola reported dipyrrin based cyclometalated Pt(II) complexes Pt-38 and Pt-39 (Fig. 22).68 The dipyrrin ligand is similar to that in BODIPY.47,48 Both complexes Pt-38 and Pt-39 show intense absorption at 503–508 nm, with ε of 18620 M−1 cm−1 and 20890 M−1 cm−1, respectively. Since the dipyrrin ligand is directly metalated by Pt(II) (in this case both dative N–Pt and covalent N–Pt bonds are formed), thus the RT phosphorescence of the dipyrrin ligand at 754−782 nm were observed, with structured emission feature, indicating the 3IL nature of the emissive triplet excited state. The RT phosphorescence wavelength are close to that observed for the BODIPY-containing Pt(II) acetylide complex (Pt-11) which shows the phosphorescence of the BODIPY moiety at 777 nm (77 K).54 RT phosphorescence lifetime of 18 ± 2 μs was observed for complex Pt-38 (phosphorescence quantum yield: 1.3%).
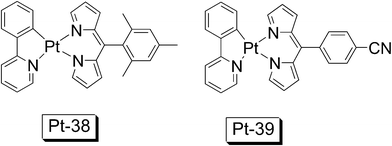 | ||
| Fig. 22 Dipyrrin based cyclometalated Pt(II) complexes Pt-38 and Pt-39 The complexes are from ref. 68. | ||
The cyclometalated Pt(II) complexes discussed in this section indicate that direct metalation of the appropriate organic chromophore is a straightforward approach to access the Pt(II) complexes that show intense absorption of visible light and the long-lived triplet excited states.
2.4 Pt(II) Schiff base complexes
Recently Pt(II) Schiff base complexes were reported to show strong absorption in visible region, compared to the typical NˆN Pt(II) acetylide complexes or cyclometalated CˆNPt(II) complexes.30 Furthermore, these complexes show moderate to high phosphorescence quantum yields, thus can be used as ideal model complexes for further derivatization to access long-lived triplet excited states.Complex Pt-40 shows absorption at 417 nm (ε = 5900 M−1 cm−1) (Fig. 23).30a Interestingly, Pt-41 shows moderate absorption at much red-shifted wavelength (ε = 9900 M−1 cm−1 at 554 nm, in benzene). Complex Pt-40 gives emissions at 550 nm/580 nm, with a phosphorescence lifetime of 3.5 μs. Conversely, Pt-41 shows red-shifted RT phosphorescence at 628 nm (in benzene) with a lifetime of 3.1 μs. With study of the zero-field splitting (ZFS) parameters, the authors proposed that the triplet excited state of the Schiff base Pt(II) complexes is ligand centered/intra-ligand charge-transfer triplet states with significant MLCT perturbations. Note that the UV-vis absorption of these complexes, especially Pt-41, are much more red-shifted than the normal CˆN cyclometalated Pt(II) OˆO complexes.2 Thus we propose that the Pt(II) Schiff based complexes (like Pt-41) are important coordination scaffolds for further functionalization.
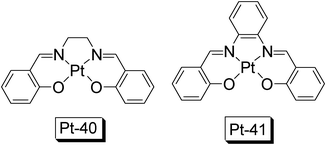 | ||
| Fig. 23 Pt(II) Schiff based complexes Pt-40 and Pt-41. The complexes are selected from ref. 30a. | ||
With the chemistry-on-complex approach, we recently prepared two pyrene-containing Pt(II) Schiff base complexes Pt-42 and Pt-43 (Fig. 24).69 The molecular design strategy is either to establish an equilibrium between the 3MLCT state and the pyrene-localized 3IL state (Pt-42), or to access the 3IL excited state (Pt-43). The two complexes were prepared with bromo-substituted Pt(II) Schiff base complexes by the Suzuki (Pt-42) or Sonogashira coupling reactions (Pt-43).
 | ||
| Fig. 24 Pyrene-containing Pt(II) Schiff base complexes Pt-42 and Pt-43. The complexes are selected from ref. 69. | ||
Pt-43
shows slightly enhanced absorption at 534 nm (ε = 13100 M−1 cm−1). Complex Pt-42 gives slightly blue-shifted absorption at 527 nm (ε = 12000 M−1 cm−1) (Fig. 25a). Different emission profiles were observed for Pt-41, Pt-42 and Pt-43 (Fig. 25b). Pt-41 and Pt-42 give similar emission bands at 614 nm and 621 nm, respectively. For Pt-43, however, a red-shifted RT phosphorescence band at 667 nm was observed. The vibrational progression of the phosphorescence band of Pt-43 is different from that of Pt-41 and Pt-42. This result indicates that the emissive triplet excited state of Pt-43 is drastically different from that of Pt-41 and Pt-42. Considering the connection profile of the pyrene unit to the Schiff base in Pt-42 and Pt-43, we propose that the emissive excited state of Pt-42 is the MLCT state, which is similar to that of Pt-41. For the complex Pt-43, however, the MLCT state is significantly perturbed by the 3IL state. Both Pt-42 and Pt-43 give much longer T1 excited state lifetime of 13.4 μs and 21.0 μs, respectively, than that of the model complex Pt-41 (τ = 4.4 μs under the same conditions).
 | ||
| Fig. 25 UV-vis absorption spectra of Pt-41, Pt-42 and Pt-43 in deaerated MeCN (1.0×10−5 M). (b) Normalized emission of Pt-41, Pt-42 and Pt-43 in MeCN solution (1.0 × 10−5 M). Reproduced from ref. 69 with permission. | ||
2.5. CˆN Cyclometalated Ir(III) complexes
Cyclometalated Ir(III) complexes are representative Ir(III) complexes that give RT phosphorescence upon photoexcitation. The typical example is complex Ir(ppy)3 (where ppy = 2-phenylpyridine). Other structural profiles have also been studied, such as Ir(ppy)2(acac), or Ir(ppy)2(bpy), etc. Note for Ir(ppy)2(bpy) the complex is positively charged.1 The model complex Ir(ppy)2(acac) (Ir-5) shows absorption at 460 nm with a very weak absorption (ε = 1600 M−1 cm−1) (Fig. 26). The emission is at 516 nm, with lifetime of 1.6 μs.1 It should be pointed out that most of the unsubstituted Ir(III) complexes show very weak absorption in visible region and the lifetime of the T1 excited state is short (less than 5 μs). | ||
| Fig. 26 Coumarin-containing Ir(III) complexes Ir-1, Ir-2, Ir-3 and Ir-4. The model complex Ir(ppy)2(acac) Ir-5 is also presented. The complexes are selected from ref. 70. | ||
In 2007, Borisov et al. reported cyclometalated Ir(III) complexes with the 7-diethylaminocoumarin as the cyclometalation ligand (Ir-1–Ir-4, Fig. 25),70 which is in the scope of direct cyclometalation of an organic fluorophore. These coumarin-containing complexes show intense absorption in the visible region beyond 450 nm. For example, Ir-4 shows ε = 92800 M−1 cm−1 at 472 nm. Efficient RT phosphorescence at 563 nm was observed (Φ = 54%). The absorption and the emission of Ir-4 are greatly red-shifted compared to that of the model complex, Ir-5. Furthermore, the lifetimes of the emissive triplet excited states of these complexes were determined to be 8.5 μs, 10.7 μs, 11.3 μs and 11.3 μs, respectively. These lifetimes are longer than the model complex Ir-5. It was proposed that the lowest-lying triplet excited states of the cyclometalated Ir(III) complexes are mixed 3MLCT/3IL states.71 These Ir(III) complexes were used for luminescent oxygen sensing. Decomposition was observed upon continuous photoexcitation.70
Dipyrrin (Dipyrrinato) was also used for preparation of cyclometalated Ir(III) complexes (Fig. 27).72Dipyrrin is the chromophore core of BODIPY dyes.47,48 Similar to cyclometalated Pt(II) complexes with the dipyrrin ligand,68 the Ir(III) complexes with dipyrrin ligand show intense absorption at ca. 485 nm (ε is up to 38400 M−1 cm−1 for Ir-6). RT phosphorescence bands with significant vibrational progression featuring in the range 650–850 nm were observed, which are attributed to the phosphorescence of the dipyrrin ligand, supported by the spin density analysis of the complexes. The lifetime of the emissive triplet excited state is up to 12.9 μs. These complexes were used for organic light emitting diodes (OLEDs), which display emission at 682 nm with maximum external quantum efficiencies up to 1.0%. It should be pointed out that the absorptions of these complexes are significantly improved compared to the model Ir(III) complexes, thus these complexes may be more significant for applications, such as photovoltaics or photocatalysis.
 | ||
| Fig. 27 Cyclometalated Ir(III) complexes with dipyrrin ligands (Ir-6 and Ir-7). The structures are from ref. 72. | ||
BODIPY was used to prepare a cyclometalated Ir(III) complex (Ir-8, Fig. 28).73Ir-8 shows strong absorption at 501 nm (ε = 83600 M−1 cm−1). Interestingly, both the fluorescence of BODIPY ligand and phosphorescence of Ir(III) coordination center were quenched, no photoluminescence was observed for Ir-8 at RT. At 77 K, the fluorescence of the BODIPY ligand at ca. 500 nm and a very weak emission band at 733 nm were observed for Ir-8. The emission band at 733 nm is attributed to the phosphorescence of the BODIPY ligand. Nanosecond time-resolved transient difference absorption spectra indicate a BODIPY localized triplet excited state, and the lifetime of the RT non-emissive triplet excited state is 25 μs.
 | ||
| Fig. 28 BODIPY-containing cyclometalated Ir(III) complex Ir-8. The complex is from ref. 73. | ||
The photophysics of Ir-8, e.g. the lack of RT luminescence, can be summarized in Fig. 29.73 Upon photoexcitation into the CT absorption band, the phosphorescence of the coordination center will be quenched by the TTET process to the 3BODIPY* excited state, which has a significantly lower energy level than the 3CT state. With excitation into the BODIPY absorption band, the ISC from 1BODIPY to the 3BODIPY quantitatively quenches the fluorescence of the BODIPY ligand, thus the complex Ir-8 is non-luminescent at RT. However, the ISC or the TTET (from Ir coordination center to the BODIPY ligand) quantum yields are unknown. It should be pointed out that the quantum yield of the 3BODIPY state is critical for the complex because a low quantum yield of the triplet excited state will be detrimental to the application of the complex, even if the complex Ir-8 shows intense absorption of visible light.
 | ||
| Fig. 29 Simplified Jablonski diagram for the photophysical process of Ir-8 upon photoexcitation into the 1CT band or the BODIPY localized absorption band. ISC is intersystem crossing and TTET is triplet–triplet energy transfer. Dashed arrows indicate non-radiative transitions. Adapted with from ref. 73 with permission. | ||
Recently we prepared coumarin-containing Ir(III) complexes, Ir-9 and Ir-10 (Fig. 30).24 The extra phenyl linker between the imidazole and the coumarin unit in Ir-10 will exclude the effect of the intramolecular hydrogen bond on the photophysical properties.24 The complex Ir-9 gives intense absorption at 466 nm (ε = 70920 M−1 cm−1). With an extra linker between the coumarin and the imidazole moiety, Ir-10 gives blue-shifted absorption at 418 nm (ε = 64170 M−1 cm−1). These absorptions are greatly enhanced compared to those of the model complexes Ir-11 and Ir-12 (Fig. 31a). Photoexcitation at the respective absorption maxima gives a drastically different emission intensity (Fig. 31b). Ir-11 and Ir-12 give intense RT phosphorescence (phosphorescence quantum yield: 55.6% and 49.2%, respectively). However, Ir-9 and Ir-10 give much weaker emission (phosphorescence quantum yields 0.6% and 0.7%, respectively).
 | ||
| Fig. 30 Coumarin-containing Ir(III) complexes Ir-9 and Ir-10. The model complexes Ir-11 and Ir-12 are also presented. The compounds are from ref. 24. | ||
 | ||
| Fig. 31 (a) UV-vis absorption of Ir-9, Ir-10, Ir-11 and Ir-12. In CH3CN (1.0×10−5 M; 20 °C). (b) Emission spectra of the Ir(III) complexes. Ir-9: λex = 462 nm, Ir-10: λex = 421 nm. Ir-11: λex = 407 nm, Ir-12: λex = 386 nm, In deaerated CH3CN (1.0 × 10−5 M; 20 °C). Reproduced from ref. 24 with permission. | ||
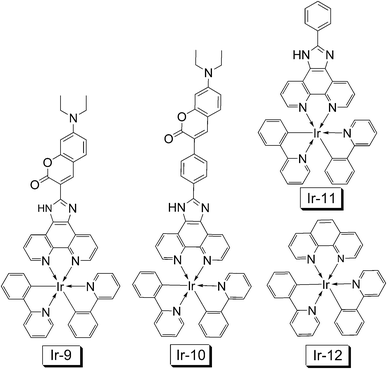
These complexes show that the intense absorption of visible light does not necessarily produce intense RT phosphorescence. However, one shouldn't be disappointed by the lack of RT emission of the Ir(III) complexes, since some triplet excited states are non-emissive at RT, but they can be produced with high efficiency upon photoexcitation of the complexes. Spin density analysis shows that 3MLCT/3IL mixed triplet excited state can be assigned for Ir-11 and Ir-12. For Ir-9 and Ir-10, however, the spin density is exclusively localized on the coumarin-imidazole ligands, and 3IL excited states can be assigned for Ir-9 and Ir-10 (Fig. 32a).
 | ||
| Fig. 32 (a) Isosurfaces of the spin density of the Ir(III) complexes Ir-9, Ir-10, Ir-11 and Ir-12 at the optimized triplet state geometry (isovalue: ±0.0004). Calculated at B3LYP/6-31G/LANL2DZ level with Gaussian 09W, based on the optimized T1 state geometry. (b) Nanosecond time-resolved transient difference absorption spectra of Ir-9. Arrows indicate the elapsed time after 355 nm laser flash. In deaerated CH3CN. 20 °C. Reproduced from ref. 24 with permission. | ||
Nanosecond time-resolved transient difference absorption spectra indicated ligand-localized triplet excited state for Ir-9 (Fig. 32b). Upon pulsed laser excitation, significant bleaching was observed at 450 nm, where the ligand gives intense absorption at the steady-state (Fig. 31). This bleaching suggests that the ground state of the ligand is drastically depleted upon photoexcitation on the time scale of μs, the most probable reason is the production of the ligand-localized long-lived triplet excited state. Similar results were observed for Ir-10.24
The lifetime of the triplet excited states of Ir-9 and Ir-10 were determined as 75.5 μs and 73.7 μs, respectively, by monitoring the decay trace of the bleaching after pulsed laser excitation (Fig. 32b). In stark contrast, much shorter lifetimes were observed for the model complexes Ir-11 and Ir-12, 0.68 μs and 0.77 μs, respectively. These lifetimes determined by the time-resolved transient difference absorption spectroscopy are very close to that obtained by the luminescence methods, thus the triplet excited state monitored by the transient difference absorption spectra are the emissive triplet excited states. However, it should be pointed out that the excited state observed with the luminescence and the transient difference absorption are not necessarily always the same.74
In this case it will be important to determine the quantum yield of the 3IL excited states of the complexes Ir-9 and Ir-10 because higher quantum yield will enhanced the photophysical processes in the applications of these complexes. The determination of the quantum yield of the 3IL triplet excited state is not straightforward, however, our TTA upconversion with the complexes Ir-9 and Ir-10 demonstrated that it is most likely that the 3IL excited states of these complexes are efficiently populated upon excitation.24
2.6 Triarylborane containing transition metal complexes
Recently, triarylborane-containing transition metal complexes have attracted much attention, due to their applications in electroluminescence and luminescent molecular probes.75–78 A review is available for the application of these complexes in optoelectronics.79 Some of these complexes show intense absorption in the visible region, as well as prolonged luminescence lifetimes.In 2011, S. Wang reported a Pt(II) complex with a triarylboron ligand that shows intense absorption in the visible region, ε = 37100 M−1 cm−1 at 456 nm.80 Note this absorption is greatly enhanced compared to the normal CˆNPt(II) acac complexes.62–64 complex Pt-44 (Fig. 33) shows intense RT phosphorescence at 590 nm (Φ = 91%). Notably, the luminescence lifetime of this complex was measured as 40.1 μs. This lifetime is significantly longer than the normal CˆN cyclometalated Pt(II) complexes.1,2,62–64 However, further detail photophysical study is left to elucidate the triplet excited state of this complex.
 | ||
| Fig. 33 Triarylborane-containing Pt-44. The complex is from ref. 80. | ||
Triarylboron-containing Ru(II) polyimine complexes Ru-8–Ru-11 (Fig. 34) were reported.81 These complexes show enhanced absorption in the visible region (ε = 21100 M−1 cm−1 for Ru-10), compared to the normal Ru(II) polyimine complexes. The emission of these complexes is red-shifted by 35 nm compared to the model complex [Ru(bpy)3]2+. The luminescence lifetimes of these complexes are longer than the usual Ru(II) polyimine complexes. For example, lifetimes of 6.2 μs, 4.2 μs, 5.8 μs and 3.5 μs were observed for Ru-8, Ru-9, Ru-10, Ru-11, respectively (at 77 K). The authors proposed that the electron-deficient boron moiety perturbed the MLCT excited state of the Ru(II) complexes. These complexes were used as molecular sensors for fluoride anions.
 | ||
| Fig. 34 Triarylboron-containing Ru(II) polyimine complexes Ru-8, Ru-9, Ru-10 and Ru-11. Mes = 2,4,6-trimethylphenyl. The complexes are from ref. 81. | ||
Two triarylboron Ru(II) complexes with similar molecular structures were reported. Interestingly, the two complexes show drastically different photophysical properties (Fig. 35).77 For example, Ru-12 shows intense absorption at 473 nm (ε = 26000 M−1 cm−1), but the analogue Ru-13 shows blue-shifted absorption at 448 nm (ε = 17000 M−1 cm−1). Furthermore, the RT phosphorescence of Ru-12 was observed at 681 nm, but Ru-13 shows much blue-shifted emission at 607 nm. The phosphorescence lifetime of Ru-12 is 12 μs, by comparison Ru-13 shows much shorter phosphorescence lifetime of 1.2 μs. By studying the interaction of the complexes with fluoride anions, the authors propose that the MLCT of the Ru(II) coordination center can be perturbed by the boron center.
 | ||
| Fig. 35 Triarylboron containing-Ru(II) polyimine complexes Ru-12 and Ru-13. The complexes are from ref. 77. | ||
3. Applications of the transition metal complexes that show intense absorption of visible light and long-lived triplet excited states
3.1 Luminescent O2 sensing
Transition metal complexes with a strong absorption of visible light and long-lived triplet excited state are important in many aspects concerning triplet–triplet energy transfer, or any other photophysical processes that required species at triplet excited state to initiate. The typical applications include luminescent O2 sensing, artificial photosynthesis, photoinduced charge separation,9 photocatalysis, TTA upconversions, etc. Some preliminary investigations have shown that the related photophysical processes can be greatly enhanced with these new complexes.28Recently we studied the effect of the lifetime of triplet excited state of Ru(II) polyimine complexes on the luminescent O2 sensing.21,28,29,82 In principle luminescent O2 sensing is a TTET process, that is, triplet–triplet energy transfer from the complex to the O2 molecules, for which the ground state is in the triplet state. According to the Stern–Volmer quenching equation, the long-lived triplet excited state is clearly beneficial for the TTET process. We used Ru-3, Ru-4 and Ru-5 for the study (Fig. 36). The O2 sensing was carried out with the complexes embedded in polymer films. We found that the quenching is drastically affected by the phosphorescence lifetime of the complexes. Ru-5 shows much significant quenching by O2 compared to that of Ru-3. Note that Ru-5 shows a lifetime of 58.4 μs and Ru-3 shows a much shorter triplet excited state lifetime of 0.45 μs. The fitting with Stern–Volmer equation gives a quenching constants of 0.35 Torr−1, which is 150-fold of that of Ru-3 (0.0023 Torr−1). This kind of enhanced luminescent O2 sensing was also observed with other complexes that show long-lived triplet excited states.21,29,70,82
 | ||
| Fig. 36 Phosphorescent intensity response of sensing films of the complexes in IMPES-C to step variations of O2 concentrations. (a), complex Ru-3, λex = 475 nm, λem = 578 nm; (b) complex Ru-5, λex = 476 nm, λem = 669 nm. Measured with home assembled optical fiber/flow cell system. The numbers indicate the O2 concentration in mixed O2/N2 gas (v/v). 25 °C. Adapted from ref. 28 with permission. | ||
3.2 Triplet–triplet annihilation (TTA) based upconversion
Recently triplet–triplet annihilation based upconversions have attracted much attention due to its advantages of readily tunable excitation/emission wavelengths, low excitation power density, efficient harvesting of the excitation light and the high upconversion quantum yields.16,18,35,83–88 For this kind of upconversion, triplet sensitizers that show intense absorption of visible light and long-lived triplet excited states are crucial. The new complexes discussed in this review are ideal candidates as triplet sensitizers for TTA upconversions.As a demonstration of the effect of visible-light harvesting and the long-lived triplet excited state on TTA upconversion, we used the cyclometalated Pt(II) complexes Pt-22, Pt-23, Pt-24 and Pt-25 (Fig. 15 and Fig. 16) as the triplet sensitizers for the TTA upconversion (Fig. 37).21 Among these complexes, Pt-23 shows the most intense absorption in visible region (Fig. 17) and the longest T1 excited state lifetime (27.9 μs in 2-methyltetrahydrofuran, 20.3 μs in dichloromethane).21,65 With 473 nm laser excitation, Pt-23 gives the most intense phosphorescence at ca. 600 nm. Other complexes give very weak emission due to the weak absorbance of these complexes at the excitation wavelength. In the presence of triplet acceptor 9,10-diphenylanthracene (DPA), the phosphorescence of Pt-23 was drastically quenched, and the upconverted fluorescence of the triplet acceptor DPA was observed at 400 nm (Fig. 37b). Other complexes show negligible upconversions under the same experimental conditions. The upconversion quantum yields (ΦUC) for Pt-23, Pt-24 and Pt-25 are 15.4%, 5.3% and 2.8%, respectively.21
![(a) Emission of Pt-22, Pt-23, Pt-24, Pt-25 and [Ru(dmb)3]2+ (1.0 × 10−5 M) with excitation by 473 nm laser (5 mW). (b) Emission of the upconverted fluorescence of 9,10-diphenylanthracene (DPA, 4.3 × 10−5 M) and the residual photoluminescence of Pt-22, Pt-23, Pt-24, Pt-25 and [Ru(dmb)3][PF6]2 (1.0 × 10−5 M) in the upconversion experiments. Excitation by 473 nm laser (5 mW). The asterisks denote the scattered laser at 473 nm. 25 °C. Reproduced from ref. 21 with permission.](/image/article/2012/RA/c1ra00665g/c1ra00665g-f37.gif) | ||
| Fig. 37 (a) Emission of Pt-22, Pt-23, Pt-24, Pt-25 and [Ru(dmb)3]2+ (1.0 × 10−5 M) with excitation by 473 nm laser (5 mW). (b) Emission of the upconverted fluorescence of 9,10-diphenylanthracene (DPA, 4.3 × 10−5 M) and the residual photoluminescence of Pt-22, Pt-23, Pt-24, Pt-25 and [Ru(dmb)3][PF6]2 (1.0 × 10−5 M) in the upconversion experiments. Excitation by 473 nm laser (5 mW). The asterisks denote the scattered laser at 473 nm. 25 °C. Reproduced from ref. 21 with permission. | ||
The efficient TTA upconversion with Pt-23 is attributed to its intense absorption of visible light, efficient ISC and the long-lived triplet excited state, all these photophysical features are important for the TTA upconversion (Fig. 37).
We also demonstrated that Ru(II) polyimine complexes with long-lived 3IL excited states are more efficient to sensitizing TTA upconversion than those with shorter T1 excited state lifetimes.20 For example, the upconversion efficiency with Ru-5 as triplet sensitizer (τT = 58.4 μs) is 10-fold of that with the model complex Ru-3 as the triplet sensitizer (τT = 0.45 μs).20
Recently we proposed that it is unnecessary for the transition metal complexes to be phosphorescent to be useful for applications in photophysical processes, such as TTA upconversion.23,24,87 This concept can be unambiguously demonstrated by the cyclometalated Ir(III) complexes Ir-9, Ir-10, Ir-11 and Ir-12 (Fig. 30).24 The upconversion quantum yield (ΦUC) of the weakly phosphorescent Ir-9 and Ir-10 was determined to be 21.3% and 23.7%, respectively. For Ir-11 and Ir-12, however, no upconversion can be observed under the same experimental conditions. Furthermore, complexes with long-lived T1 excited states (Ir-9 and Ir-10) give a much large Stern–Volmer quenching constant, an indication of the efficient TTET process.
The detailed discussion of the application of transition metal complexes that show intense absorption of visible light and long-lived T1 excited state in TTA upconversions has been summarized in a recent review.88
We believe that the application of transition metal complexes that show intense absorption of visible light and long-lived triplet excited states is still in its infancy and much room is left for further explorations in areas such as photovoltaics,89 photocatalysis,90,91 luminescent molecular probes,14,15,92,93etc.
4. Conclusions and outlook
Transition metal complexes, such as with Pt(II), Ir(III) and Ru(II) coordination centers, with strong absorption of visible light and long-lived triplet excited states are useful for applications such as photovoltaics, photocatalysis, luminescent molecular probes and upconversion, etc. These photophysical features are drastically different from the traditional applications of these complexes, such as electroluminescence, for which the strong absorption of visible light is unnecessary and in stark contrast to the complexes discussed in this review article, the lifetime of the triplet excited state of the complexes for electroluminescence must be short to avoid the saturation effect. Thus, new molecular design rationales have to be developed for new applications of the complexes. Herein we summarized the recently reported examples and proposed some preliminary rules for the design of such complexes, such as direct metalation of an organic chromophore that shows strong absorption of visible light, such as coumarin, naphthalimide (NI), naphthalenediimide (NDI), etc. With this strategy, complexes with the desired features of strong absorption of visible light and long-lived T1 excited state can be obtained. At the same time, relative energy levels of excited states of the dyad components have to be considered in design of the complexes. The current examples also indicated some elusive photophysics of these transition metal–organic chromophore dyads, for example, complexes with intense absorption do not produce any RT emission. In some cases, however, the lack of the RT emission of the complexes does not deter the complexes from applications, as long as the triplet excited state can be efficiently populated upon photoexcitation. The methods used in the study of long-lived triplet excited states, such as nanosecond time-resolved transient difference absorption spectroscopy and the DFT calculations (spin density analysis, etc), are also discussed. Some preliminary applications of the transition metal complexes that show strong visible-light absorption and long-lived triplet excited states have been presented, such as for luminescent O2 sensing and as triplet sensitizers for triplet–triplet annihilation (TTA) upconversions. It has been proved that these photophysical processes were greatly enhanced with the title complexes. It should be pointed out that either the preparation or the application of the transition metal complexes that show strong visible light absorption and long-lived triplet excited states are still in the early age, for example, no applications of these complexes for photovoltaics or photocatalysis have been reported. Thus we believe that much room is left for the development of this fascinating area, for which synthetic chemistry, photophysics and photochemistry can be combined to address the challenges of design and application of these interesting compounds.Acknowledgements
We thank the NSFC (20972024, 21073028 and 21103015), the Royal Society (UK) and NSFC (China-UK Cost-Share Science Networks, 21011130154), the Fundamental Research Funds for the Central Universities (DUT10ZD212 and DUT11LK19) and Ministry of Education (SRFDP-200801410004 and NCET-08-0077) for financial support. Last but not the least, we are grateful to all the colleagues around the world for their enthusiastic contributions to this fascinating research area.References
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigellet, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS.
- J. A. Gareth Williams, Top. Curr. Chem., 2007, 281, 205 CrossRef.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. V. Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117 CrossRef CAS.
- X. –Y. Wang, A. D. Guerzo and R. H. Schmehl, J. Photochem. Photobiol., C, 2004, 5, 55 CrossRef CAS.
- E. A. Medlycott and G. S. Hanan, Coord. Chem. Rev., 2006, 250, 1763 CrossRef CAS.
- N. Armaroli, ChemPhysChem, 2008, 9, 371 CrossRef CAS.
- (a) F. Gao, Y. Wang, D. Shi, J. Zhang, M. Wang, X. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2008, 130, 10720 CrossRef CAS; (b) M. Borgström, N. Shaikh, O. Johansson, M. F. Anderlund, S. Styring, B. A kermark, A. Magnuson and L. Hammarström, J. Am. Chem. Soc., 2005, 127, 17504 CrossRef; (c) B. Geiß and C. Lambert, Chem. Commun., 2009, 1670 Search PubMed; (d) S. Suzuki, R. Sugimura, M. Kozaki, K. Keyaki, K. Nozaki, N. Ikeda, K. Akiyama and K. Okada, J. Am. Chem. Soc., 2009, 131, 10374 CrossRef CAS.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596 CrossRef.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS.
- A. G. Condie, J. C. Gonzalez-Gomez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS.
- J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368 CrossRef CAS.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS.
- Y. Yap Cheng, B. Fckel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795 CrossRef.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626 CrossRef CAS.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Dalton Trans., 2011, 40, 5953 RSC.
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834 RSC.
- (a) Y. Liu, W. Wu, J. Zhao, X. Zhang and H. Guo, Dalton Trans., 2011, 40, 9085–9089 RSC; (b) L. Huang, L. Zeng, H. Guo, W. Wu, W. Wu, S. Ji and J. Zhao, Eur. J. Inorg. Chem., 2011, 4527–4533 CrossRef CAS; (c) W. Wu, H. Guo, J. Zhao, J. Sun, S. Ji and Z. Wang, Chem.–Eur. J., 2011 Search PubMed , chem.201102634. In press..
- J. Sun, W. Wu, H. Guo and J. Zhao, Eur. J. Inorg. Chem., 2011, 3165 CrossRef CAS.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447 CrossRef CAS.
- C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053 CrossRef CAS.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819 CrossRef CAS.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo, P. Song, K. Han, L. Chi, J. Shao and J. Zhao, J. Mater. Chem., 2010, 20, 9775 RSC.
- (a) C.–M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem.–Eur. J., 2010, 16, 233 CrossRef CAS; (b) P. Wu, D.–L. Ma, C.–H. Leung, S.–C. Yan, N. Zhu, R. Abagyan and C.–M. Che, Chem.–Eur. J., 2009, 15, 13008 CrossRef CAS.
- N. D. McClenaghan, Y. Leydet, B. Maubert, M. T. Indelli and S. Campagna, Coord. Chem. Rev., 2005, 249, 1336 CrossRef CAS.
- A. I. Baba, J. R. Shaw, J. A. Simon, R. P. Thummel and R. H. Schmehl, Coord. Chem. Rev., 1998, 171, 43 CrossRef CAS.
- D. B. Papkovsky and T. C. O'Riordan, J. Fluoresc., 2005, 15, 569 CrossRef CAS.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. Lett., 2010, 1, 195 CrossRef CAS.
- M. Abrahamsson, M. Jager, R. J. Kumar, T. Osterman, P. Persson, H. –C. Becker, O. Johansson and L. Hammarstrom, J. Am. Chem. Soc., 2008, 130, 15533 CrossRef CAS.
- D. V. Kozlov, D. S. Tyson, C. Goze, R. Ziessel and F. N. Castellano, Inorg. Chem., 2004, 43, 6083 CrossRef CAS.
- C. Goze, D. V. Kozlov, D. S. Tyson, R. Ziessel and F. N. Castellano, New J. Chem., 2003, 27, 1679 RSC.
- G. J. Wilson, A. Launikonis, W. H. F. Sasse and A. W.-H. Mau, J. Phys. Chem. A, 1997, 101, 4860 CrossRef CAS.
- D. S. Tyson and N. Castellano Felix, J. Phys. Chem. A, 1999, 103, 10955 CrossRef CAS.
- D. S. Tyson, J. Bialecki and F. N. Castellano, Chem. Commun., 2000, 2355 RSC.
- A. D. Guerzo, S. Leroy, F. Fages and R. H. Schmehl, Inorg. Chem., 2002, 41, 359 CrossRef.
- A. F. Morales, G. Accorsi, N. Armaroli, F. Barigelletti, S. J. A. Pope and M. D. Ward, Inorg. Chem., 2002, 41, 6711 CrossRef CAS.
- A. Harriman, A. Khatyr and R. Ziessel, Dalton Trans., 2003, 2061 RSC.
- Q.–Z. Yang, L.–Z. Wu, Z.–X. Wu, L.–P. Zhang and C.–H. Tung, Inorg. Chem., 2002, 41, 5653 CrossRef CAS.
- B. Maubert, N. D. McClenaghan, M. T. Indelli and S. Campagna, J. Phys. Chem. A, 2003, 107, 447 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611 RSC.
- M. Galletta, F. Puntoriero, S. Campagna, C. Chiorboli, M. Quesada, S. Goeb and R. Ziessel, J. Phys. Chem. A, 2006, 110, 4348 CrossRef CAS.
- X. Han, L.–Z. Wu, G. Si, J. Pan, Q.–Z. Yang, L.–P. Zhang and C.–H. Tung, Chem.–Eur. J., 2007, 13, 1231 CrossRef CAS.
- I. E. Pomestchenko, C. R. Luman, M. Hissler, R. Ziessel and F. N. Castellano, Inorg. Chem., 2003, 42, 1394 CrossRef CAS.
- P.–H. Lanoë, J.–L. Fillaut, L. Toupet, J. A. G. Williams, H. L. Bozec and V. Guerchais, Chem. Commun., 2008, 4333 RSC.
- A. A. Rachford, S. Goeb, R. Ziessel and F. N. Castellano, Inorg. Chem., 2008, 47, 4348 CrossRef CAS.
- F. Nastasi, F. Puntoriero, S. Campagna, S. Diring and R. Ziessel, Phys. Chem. Chem. Phys., 2008, 10, 3982 RSC.
- A. A. Rachford, S. Goeb and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 2766 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. Gareth Williams, Inorg. Chem., 2011, 50, 3804 CrossRef CAS.
- Z. Xu, X. Qian and J. Cui, Org. Lett., 2005, 7, 3029 CrossRef CAS.
- J. Huang, Y. Xu and X. Qian, Org. Biomol. Chem., 2009, 7, 1299 CAS.
- D. S. Tyson, C. R. Luman, X. Zhou and F. N. Castellano, Inorg. Chem., 2001, 40, 4063–4071 CrossRef CAS.
- G. J. Ryan, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2008, 47, 401 CrossRef CAS.
- H. Guo, M. L. Muro-Small, S. Ji, J. Zhao and F. N. Castellano, Inorg. Chem., 2010, 49, 6802 CrossRef CAS.
- G.–J. Zhou, Q. Wang, W.–Y. Wong, D. Ma, L. Wang and Z. Lin, J. Mater. Chem., 2009, 19, 1872 RSC.
- Z. He, W. –Y. Wong, X. Yu, H. –S. Kwok and Z. Lin, Inorg. Chem., 2006, 45, 10922 CrossRef CAS.
- B. Yin, F. Niemeyer, J. A. G. Williams, J. Jiang, A. Boucekkine, L. Toupet, H. L. Bozec and V. Guerchais, Inorg. Chem., 2006, 45, 8584 CrossRef CAS.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS.
- J. Hu, J. H. K. Yip, D.–L. Ma, K.–Y. Wong and W.–H. Chung, Organometallics, 2009, 28, 51 CrossRef CAS.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Eur. J. Inorg. Chem., 2010, 4470 CrossRef CAS.
- C. Bronner, S. A. Baudron, M. W. Hosseini, C. A. Strassert, A. Guenetb and L. D. Cola, Dalton Trans., 2010, 39, 180 RSC.
- W. Wu, J. Sun, S. Ji, W. Wu, J. Zhao and H. Guo, Dalton Trans., 2011, 40, 11550 RSC.
- S. M. Borisov and I. Klimant, Anal. Chem., 2007, 79, 7501 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. –E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
- K. Hanson, A. Tamayo, V. V. Diev, M. T. Whited, P. I. Djurovich and M. E. Thompson, Inorg. Chem., 2010, 49, 6077 CrossRef CAS.
- A. A. Rachford, R. Ziessel, T. Bura, P. Retailleau and F. N. Castellano, Inorg. Chem., 2010, 49, 3730 CrossRef CAS.
- D. S. Tyson, K. B. Henbest, J. Bialecki and F. N. Castellano, J. Phys. Chem. A, 2001, 105, 8154 CrossRef CAS.
- Z. M. Hudson, C. Sun, M. G. Helander, H. Amarne, Z.–H. Lu and S. Wang, Adv. Funct. Mater., 2010, 20, 3426 CrossRef CAS.
- W.–J. Xu, S.–J. Liu, X.–Y. Zhao, S. Sun, S. Cheng, T.–C. Ma, H.–B. Sun, Q. Zhao and W. Huang, Chem.–Eur. J., 2010, 16, 7125 CAS.
- E. Sakuda, Y. Ando, A. Ito and N. Kitamura, Inorg. Chem., 2011, 50, 1603 CrossRef CAS.
- Z. M. Hudson, S.–B. Zhao, R.–Y. Wang and S. Wang, Chem.–Eur. J., 2009, 15, 6131 CrossRef CAS.
- Z. M. Hudson and S. Wang, Dalton Trans., 2011, 40, 7805 RSC.
- Z. M. Hudson, M. G. Helander, Z.–H. Lu and S. Wang, Chem. Commun., 2011, 47, 755 RSC.
- Y. Sun, Z. M. Hudson, Y. Rao and S. Wang, Inorg. Chem., 2011, 50, 3373 CrossRef CAS.
- H. Guo, S. Ji, W. Wu, W. Wu, J. Shao and J. Zhao, Analyst, 2010, 135, 2832 RSC.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776 RSC.
- H.–C. Chen, C.–Y. Hung, K.–H. Wang, H.–L. Chen, W. S. Fann, F.–C. Chien, P. Chen, T. J. Chow, C.–P. Hsu and S.–S. Sun, Chem. Commun., 2009, 4064 RSC.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinar, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef.
- P. Du and R. Eisenberg, Chem. Sci., 2010, 1, 502 RSC.
- S. Ji, H. Guo, W. Wu, W. Wu and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 8283 CrossRef CAS.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937 RSC.
- A. Abbotto, C. Barolo, L. Bellotto, F. D. Angelis, M. Grätzel, N. Manfredi, C. Marinzi, S. Fantacci, J.–H. Yum and M. K. Nazeeruddin, Chem. Commun., 2008, 5318 RSC.
- F. Wang, W.–G. Wang, X.–J. Wang, H.–Y. Wang, C.–H. Tung and L.–Z. Wu, Angew. Chem., Int. Ed., 2011, 50, 3193 CrossRef CAS.
- (a) J. Zhang, P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2007, 129, 7726 CrossRef CAS; (b) X. Wang, S. Goeb, Z. Ji, N. A. Pogulaichenko and F. N. Castellano, Inorg. Chem., 2011, 50, 705 CrossRef CAS; (c) Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056 CrossRef CAS.
- P.–H. Lanöe, H. L. Bozec, J. A. G. Williams, J.–L. Fillaut and V. Guerchais, Dalton Trans., 2010, 39, 707 RSC.
- S. Ji, H. Guo, X. Yuan, X. Li, H. Ding, P. Gao, C. Zhao, W. Wu, W. Wu and J. Zhao, Org. Lett., 2010, 12, 2876 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |