DOI:
10.1039/C2RA00445C
(Paper)
RSC Adv., 2012,
2, 7066-7073
Hindered rotation in a novel 1,2,4-triazinyl phenanthroline (t-phen) ligand leading to improved separation of Am3+ and Eu3+vis-à-vis 1,2,4-triazinyl bipyridine (t-bipy): a computational validation of the experimental results†
Received
13th July 2011
, Accepted 27th May 2012
First published on 6th July 2012
Abstract
A novel nitrogen donor ligand having a more pre-organized structure, 1,2,4-triazinyl phenanthroline (t-phen), is synthesized and evaluated for lanthanide–actinide separation in the present work. The extraction and selectivity for Am3+ over Eu3+ was found to be improved with t-phen as compared to the analogous 1,2,4-triazinyl bipyridine (t-bipy), which was explained by analyzing their conformational energies and energy differences between the frontier orbitals (highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO)) with the help of density functional theoretical calculations. Higher covalence in Am3+ complexes as compared to the Eu3+ complexes was indicated by more shared electrons between Am3+ and bonded ‘N’ atoms, shorter ‘Am–N’ bonds and higher overlap between the orbitals of Am3+ and ligands in the frontier orbitals of the complexes in the case of both the ligands. Natural bond order analysis also supports these observations.
Introduction
The high level waste (HLW) generated from the PUREX process contains a number of fission products, including several lanthanide elements along with unextracted uranium, plutonium and very small amount of minor actinides, i.e. Am and Cm. The major concern for the safe management of HLW is due to the presence of minor actinides because of their long half lives and high radiotoxicity. The safe management of these minor actinides is possible either by vitrifying them in a proper solid matrix or by their transmutation into short lived or stable nuclides. For this purpose, the minor actinides need to be separated from the bulk of the metal ions present in the HLW. In the process called “Actinide Partitioning” the minor actinides are extracted along with the lanthanides leaving behind most of the other elements in the raffinate. The co-extraction of the trivalent lanthanides and actinides is a consequence of their similar chemical behaviour.1 However, the transmutation process using high flux reactors necessitates the separation of trivalent actinides from the trivalent lanthanides which act as neutron poisons. The current strategy on lanthanide–actinide separation is based on the use of soft donor extractants for preferential actinide extraction. The higher spatial distribution of ‘5f’ orbitals of the actinides as compared to the ‘4f’ orbitals of the lanthanides leads to the formation of preferential covalent bonding of actinides with soft donor ligands as compared to that of lanthanides and this property is being exploited for the separation of trivalent actinides and lanthanides. A number of ‘N’ donor heteropolycyclic ligands starting from terpyridine (terpy) (Fig. 1a) to bis-1,2,4-triazinyl pyridine (BTP) (Fig. 1b) and bis-1,2,4-triazinyl bipyridine (BTBP) (Fig. 1c) have been evaluated for this purpose.2,3 The complexes formed with Am3+ or Ln3+ are very different for different ‘N’ donor ligands, viz. the terpy complexes of Ln3+ or An3+ contain only one or two terpy molecules4,5 whereas three molecules of BTP are present6,7 in An3+/Ln3+ BTP complexes. Hudson et al., therefore, explored a class of ligands, i.e. 6-(5,6-dialkyl-1,2,4-triazine-3-yl) bipyridine (R2(t-bipy)) (Fig. 1d) derivatives which fall in between the terpy and BTP ligands.8 They found that only one t-bipy molecule is present in the extractable complexes of Ln3+ and An3+, which resembles the behaviour of terpy. Poor extraction and selectivity for trivalent actinides over the lanthanides was observed employing the t-bipy derivatives as the extractant. Hudson et al. analyzed the energies of different conformations of free t-bipy molecules and observed that the rotation around the two pyridine rings is much more hindered (ΔE ∼ 9.0 kcal mol−1) as compared to the pyridine-triazine rings (ΔE ∼ 0.78 kcal mol−1), because of the presence of ortho-hydrogen atoms in the two pyridine rings in the cis conformation. The requirement of this 9.0 kcal mol−1 energy can be avoided if we can preorganize the two ‘N’ atoms of the pyridine rings in a cis conformation resulting in the formation of a stronger complex with the metal ion as compared to t-bipy derivatives. An attempt was, therefore, made in the present work to synthesize a ligand where the 1,2,4-triazine ring is attached to the 1,10-phenanthroline ring (2-(5,6-dialkyl-1,2,4-triazine-3-yl)1,10-phenanthroline (R2(t-phen))) (Fig. 1e) instead of the bipyridine ring in R2(t-bipy). The extraction and separation behaviour of Am3+ and Eu3+ with Me2(t-bipy) and Me2(t-phen) was compared at different pH values of the aqueous phase. Computational studies were carried out to corroborate the higher Am3+ extraction and selectivity of t-phen as compared to that of t-bipy.
 |
| | Fig. 1 Various ‘N’ donor heteropolycyclic ligands for the separation of trivalent actinides and lanthanides; (a) terpy, (b) BTP, (c) BTBP (d) t-bipy and (e) t-phen. | |
Experimental
Synthesis of Me2(t-phen)
a) 1,10-Phenanthroline-1-oxide.
Hydrogen peroxide (4 ml, 30% solution in water) was added to a solution of 1,10-phenanthroline monohydrate (5 g, 25.25 mmol) in glacial acetic acid (30 ml). The reaction mixture was stirred at 75 °C for 4 h and another portion of hydrogen peroxide (4 ml, 30% solution in water) was added to the mixture. After 3 h at 75 °C, the reaction mixture was cooled to room temperature and acetic acid was neutralized slowly by adding solid sodium carbonate into the reaction mixture. The resultant pasty mass was extracted multiple times with chloroform. The combined extract was concentrated to give a yellow green colored solid that was crystallized from chlorobenzene to give 1,10-phenanthroline-1-oxide (4.16 g, 84%).9 M.P. 184–185 °C, (reported M.P. 180–181 °C) IR (KBr): 3065, 1626, 1598, 1577, 1517, 1412, 1327, 1256, 1203, 1069, 1016, 842, 815, 711, 679 cm−1; 1H NMR (200 MHz, CDCl3): δ 7.39 (dd, 1 H, J = 6.4, 8.0 Hz), 7.57–7.761 (m, 4 H), 8.17 (dd, 1 H, J = 1.8, 8 Hz), 8.68 (d, 1 H, J = 6.4 Hz), 9.25 (dd, 1 H, J = 1.8, 4.3 Hz); 13C NMR (50 MHz, CDCl3): δ 122.5, 122.8, 123.2, 124.2, 126.1, 128.5, 128.6, 132.9, 135.5, 140.3, 142.2, 149.4.
b) 2-Cyano-1,10-phenanthroline.
Freshly distilled benzoyl chloride (4.75 ml, 40.9 mmol) was added slowly (about 15 min) to a vigorously stirred solution of 1,10-phenanthroline-1-oxide (5 g, 25.51 mmol) and potassium cyanide (4.72 g, 72.42 mmol) in distilled water (80 ml) at room temperature. After 15 min, the solid formed was collected by filtration, washed with water and dried in vacuuo. Light tan needles were obtained after crystallization from ethanol (3.92 g, 75%).9
M.P. 234–235 °C, lit.1 M.P. 233–234 °C. IR (KBr): 3068, 2229, 1505, 1486, 1390 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.76 (dd, 1 H, J = 4.5, 8.1 Hz), 7.87 (d, 1 H, J = 9 Hz), 7.98 (d, 1 H, J = 9 Hz), 7.99 (d, 1 H, J = 8.1 Hz), 8.33 (dd, 1 H, J = 1.5, 8.1 Hz), 8.42 (d, 1 H, J = 8.1 Hz), 9.3 (dd, 1 H, J = 1.8 Hz, 4.2 Hz). 13C NMR (50 MHz, CDCl3): δ 117.2, 123.9, 125.5, 126.0, 129.0, 129.6, 133.2, 136.1, 137.0, 145.0, 146.5, 151.0. MS (ESI) m/z: 205 (M+, 100%), 178 (15), 152 (12), 125 (9), 102 (9), 89 (3), 75 (3). GC (260 °C isothermal): tR 21.6 min (100%).
c) 3-{2-(1,10-Phenanthrolyl) -(5,6-dimethyl)}-1,2,4-triazine (Me2(t-phen)).
Hydrazine hydrate (7 ml, 224 mmol) was added to a solution of 2-cyano-1,10-phenanthroline (2.5 g, 12.2 mmol) in ethanol (85 ml) and stirred overnight. The reaction mixture was concentrated under vacuum and the residue was crystallised from water to give the hydrazone (2.38 g, 82%). A mixture of monocarboxamide hydrazone (2.0 g, 8.44 mmol) and butan-2,3-dione (0.85 ml, 9.54 mmol) in benzene (13 ml) was heated under reflux. After 2.5 h, the solvent was evaporated and the residue crystallized from ethanol to give the hemi triazinyl monohydrate derivative (2.1 g, 82%).10 M.P. 211–212 °C, lit.10 M.P. 210–211 °C. 1H NMR (200 MHz, CDCl3): δ 2.79 (s, 3 H), 2.80 (s, 3 H), 3.90 (s, broad, 2 H, H2O), 7.69(dd, 1 H, J = 4.4, 8 Hz), 7.87 (s, 2 H), 8.30 (dd, 1 H, J = 1.4, 8 Hz), 8.45 (d, 1 H, J = 8.4 Hz), 8.98 (d, 1 H, J = 8.4 Hz), 9.22 (dd, 1 H, J = 1.4, 4.4 Hz). 13C NMR (50 MHz, CDCl3): δ 19.5, 21.8, 122.7, 123.2, 126.2, 127.8, 128.9, 129.4, 136.1, 137.0, 146.1, 146.2, 150.3, 152.8, 157.2, 159.8, 161.6.
Synthesis of Me2(t-bipy)
a) 2,2′-Bipyridine-N-oxide.
A solution of 3-chloroperoxybenzoic acid (3.2 g, 70%, 12.9 mmol) in dichloromethane (22.8 ml) was added drop wise to a solution of bipyridine (2 g, 12.6 mmol) in dichloromethane (13.4 ml) at 5–10 °C. After 16 h at room temperature, the reaction mixture was cooled on an ice bath and sodium hydroxide (2 M solution in water, 14.5 ml, 29 mmol) was added into it. After 15 min, the reaction mixture was diluted with chloroform (25 ml). The organic layer was separated and washed with 2 M NaOH solution, dried over sodium carbonate and evaporated under reduced pressure. The residue was purified by silica gel column chromatography to give the mono N-oxide (1.7 g, 78%).11 M.P. 54–55 °C (reported M.P. 55–56 °C), 1H NMR (200 MHz, CDCl3): δ 7.27–7.42 (m, 3 H), 7.84 (dt, 1 H, J = 1.8, 8 Hz), 8.19 (dd, 1 H, J = 2, 8 Hz), 8.33 (d, 1 H, J = 6.2), 8.71 (d, 1 H, J = 4.7 Hz), 8.87 (d, 1 H, J = 8 Hz). 13C NMR (50 MHz, CDCl3): δ 124.0, 125.0, 125.2, 127.6, 136.0, 140.4, 147.0, 149.0, 149.4.
b) 2-Cyano-2,2′-bipyridine.
A solution of 2,2′-bipyridine-N-oxide (0.3 g, 1.7 mmol) and trimethylsilyl cyanide (0.27 ml, 1.88 mmol) in dichloromethane (3.5 ml) was stirred for 5 min at room temperature. N,N′- dimethylcarbamoyl chloride (0.17 ml, 1.88 mmol) was added to the reaction mixture and stirred for 7 days under an argon atmosphere. The reaction mixture was diluted with diethyl ether (30 ml) and washed successively with 5% NaHCO3 solution and brine and dried over solid Na2CO3. The solvent was evaporated and the residue was purified by silica gel column chromatography to give the monocyano compound (0.29 g, 93%).11
M.P. 132–133 °C (reported M.P. 132–133 °C), 1H NMR (200 MHz, CDCl3): δ 7.37 (ddd, 1 H, J = 1.1, 4.8, 7.5 Hz), 7.70 (dd, 1 H, J = 1, 7.5 Hz), 7.85 (dt, 1 H, J = 1.8, 8 Hz), 7.94 (t, 1 H, J = 8 Hz), 8.46 (d, 1 H, J = 8.0 Hz), 8.64–8.69 (m, 2 H). 13C NMR (50 MHz, CDCl3): δ 117.3, 121.5, 124.2, 124.7, 128.0, 133.2, 137.2, 137.8, 149.2, 153.9, 157.6.
c) 6-(5,6-Dimethyl-1,2,4-triazin-3yl)-2,2′-bipyridine (Me2(t-bipy)).
Hydrazine hydrate (0.19 ml, 6 mmol) was added to a solution of 6-cyano-2,2′-bipyridine (0.108 g, 0.6 mmol) dissolved in ethanol (7.2 ml) and stirred for 2 days. The solvent was removed under reduced pressure to give the monocarboxamide hydrazone (0.133 g, 0.6 mmol, 100%). Butan-2,3-dione (0.06 g, 0.7 mmol) and benzene (2 ml) were added to the monocarboxamide hydrazone and the mixture was refluxed for 5 h. The solvent was evaporated and the residue was purified by silica gel column chromatography to give the Me2(t-bipy)8 (0.117 g, 74%).8 M.P. 148–149 °C. 1H NMR (200 MHz, CDCl3): δ 2.68 (s, 3 H), 2.77 (s, 3 H), 7.38 (t, 1 H, J = 5.4 Hz), 7.91 (t, 1 H, J = 7.6 Hz), 8.03 (t, 1H, J = 7.9 Hz), 8.56–8.73 (m, 4 H). 13C NMR (50 MHz, CD3OD): δ 19.5, 21.9, 123.3, 123.7, 124.8, 125.5, 138.8, 139.3, 149.8, 153.6, 156.6, 157.3,158.9, 161.9, 162.2. Calcd. for C15H13N5; C, 68.42; H, 4.98; N, 26.60%; Found C, 67.93; H, 5.27; N, 26.36%.
Other reagents and chemicals
2-bromo-octanoic acid (>97%) was procured from Fluka chemie, Germany. 241Am was purified as reported earlier12 and the purity was checked by alpha spectrometry. 152,154Eu was procured from the Board of Radiation and Isotope Technology (BRIT), Mumbai, India. Suprapur nitric acid (Merck) was used for preparing the tracer solutions. All other reagents were of AR grade.
Distribution studies
Distribution studies were carried out using 241Am and 152,154Eu tracers under varying experimental conditions. A mixture of 0.03 M Me2(t-bipy) or Me2(t-phen) and 1 M 2-bromo-octanoic acid in n-dodecane was used as the organic phase while the aqueous phase was the dilute nitric acid in the pH range of 1.2 to 2.2. Equal volumes (1.0 mL) of the organic and aqueous phases containing the required tracer were kept for equilibration in a thermostated water bath at 25 ± 0.1 °C for 60 min. The two phases were then centrifuged and assayed by taking suitable aliquots (100–200 μL) from both the phases. The gamma activities were measured using a high purity germenium detector procured from Baltic Scientific Instruments. The distribution ratio (DM) was calculated as the ratio of counts per minute per unit volume in the organic phase to that in the aqueous phase. The mass balance was within the experimental error limits (± 5%). The separation factor (S.F.) was calculated as the ratio of DAm to DEu.
Computational studies
The alkyl substituents show insignificant changes in the charge distributions and the energy gap between the frontier orbitals.13 A number of studies were also carried out, where H-form of BTP was considered as a model for the alkyl BTP derivatives.14,15 Most of the calculations of the free ligands and their Am3+ and Eu3+ complexes were, therefore, performed for hydrogen form of both the ligands (t-bipy and t-phen). The presence of alkyl groups can, however, alter the conformational energies require to rotate the rings. The relative energies of different conformations of Me2(t-bipy) and Me2(t-phen) were determined varying the angle of rotation around the two adjacent rings. All the calculations of free and protonated ligands were performed at the density functional (DFT) level of theory using the hybrid exchange correlation functional (B3LYP) with the GAMESS molecular orbital package16 using the valence double zeta Gaussian type of basis set 6-31G(d,p). Molecular orbital pictures were drawn using the visualizing program MOLDEN.17 The calculations on the Am3+ and Eu3+ complexation were carried out with TURBOMOLE program package18 at the DFT level using Becke′s exchange functional19 in conjunction with Perdew′s correlation functional20 (BP86) where for the heavy atoms relativistic energy consistent ab initio f-in-valence 28 (Eu) and 60 (Am) electron core pseudo potentials (ECP) along with the corresponding def-SV(P) basis set were selected. All other lighter atoms were treated at the all electron (AE) level and the standard def-SV(P) basis sets as implemented in the TURBOMOLE program was used. It may be noted that for the Am and Eu atoms the def-SV(P) basis set as present in the TURBOMOLE basis set library is quite large and consists of (14s13p10d8f1g) functions contracted to [10s9p5d4f1g].21–27
Results and discussion
Distribution studies of Am3+ and Eu3+
The distribution behaviour of Am3+ and Eu3+ by Me2(t-bipy) and Me2(t-phen) was compared in the presence of 2-bromo-octanoic acid in an n-dodecane medium at different aqueous phase pHs. With increasing the pH of the aqueous phase, the distribution ratio (DM) of both Am3+ and Eu3+ was found to increase with similar slopes due to more deprotonation of 2-bromo-octanoic acid with increasing pH of the aqueous phase (Fig. 2). When the extraction efficiency of these two ligands for Am3+ over Eu3+ was compared, it was interesting to note that the DAm values were an order of magnitude higher in case of Me2(t-phen) as compared to Me2(t-bipy) at all pHs (Fig. 2). Beside the DAm value, selectivity for Am3+ over Eu3+ was also found to be higher with Me2(t-phen) as compared to Me2(t-bipy) (Fig. 3). An attempt was, therefore made in order to rationalize these observations with the help of computational studies.
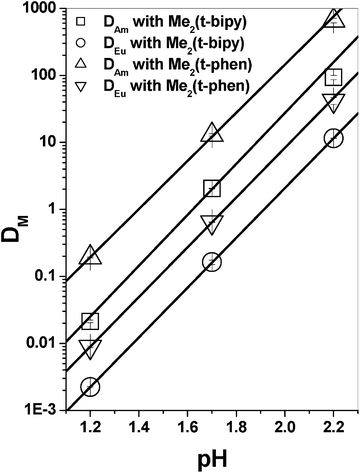 |
| | Fig. 2 The effect of aqueous phase pH on the extraction of Am3+ and Eu3+ with Me2(t-phen) and Me2(t-bipy); Org. Phase: 0.03 M ligand + 1 M 2-bromo-octanoic acid in n-dodecane; Aq. Phase: dilute HNO3 with varying pH. | |
 |
| | Fig. 3 A comparison of the selectivity of Am3+ over Eu3+ with the two ligands (Me2(t-phen) and Me2(t-bipy)); Org. Phase: 0.03 M ligand + 1 M 2-bromo-octanoic acid in n-dodecane; Aq. Phase: dilute HNO3 with varying pH. | |
Computational studies of free ligands
Analysis of conformational energies of Me2(t-bipy) and Me2(t-phen).
During the geometry optimization of Me2(t-bipy) and Me2(t-phen), the lowest energy (global minimum) was achieved in the trans–trans (tt) and trans (t) conformations, respectively. A number of local minima are, however, possible for this class of molecules. A local minimum was obtained with non co-planar geometry of the aromatic rings when the rings are in cis-conformation. In the case of Me2(t-bipy), the dihedral angle is as high as 37.8° between the two pyridine rings and 10° between the central pyridine-triazine rings at the local minimum. For the free 2,2′bipyridine molecule, similarly, the global minimum at a dihedral angle of 180° (the trans planar conformation) and a local minimum at around 45° were reported.28 The higher dihedral angle between the two pyridine rings is due to the unfavourable interaction between the ortho-hydrogen atoms of the pyridine rings which is much lower in the case of pyridine-triazine rings as the triazine ring does not contain any ortho-hydrogen atoms. There are four possible low energy conformations (minima) for Me2(t-bipy) (tt, tc, ct and cc (Fig. 4)), which is described in detail in the literature .8 The lowest energy conformation is ‘tt’, but the conformation ‘tc’ is only 0.7 kcal mol−1 higher in energy. The relative energies of the conformations ‘ct’ (7.4 kcal mol−1) and ‘cc’ (8.9 kcal mol−1) are higher in energy as compared to the ‘tt’ and ‘tc’ conformations. The reason for the higher conformational energies of the ‘cc’ and ‘ct’ conformations is the presence of ortho-hydrogen atoms in the two pyridine rings of t-bipy in the ‘cc’ and ‘ct’ conformations, which makes the two pyridine rings non coplanar. The ligand needs to be in the ‘cc’ conformation for the complexation with metal ions. There are two possible ways to convert Me2(t-bipy) from the most stable ‘tt’ to the complexable ‘cc’ conformation, either through ‘ct’ or through ‘tc’ conformations (Fig. 4). The route through the ‘tc’ conformation is energetically more favourable because of the lower energy of the intermediate ‘tc’ as compared to the ‘ct’ conformation. The transition states (TS) between various minima were also found out. The lesser heights of the two energy barriers in the ‘tc’ route (relative energies of TS are 5.6 and 10.2 kcal mol−1) as compared to that in the ‘ct’ route (relative energies of TS are 8.8 and 12.6 kcal mol−1) may also favour the ‘tc’ route. Me2(t-bipy), however, has to cross two energy barriers through any of the two routes (Fig. 4). In the case of the ligand Me2(t-phen), only two low energy conformations (cis and trans) are possible (Fig. 5). The ‘cis’ conformation is higher in energy than its most stable trans-conformation by only 0.5 kcal mol−1 and they are interconvertable through a single barrier of much lower energy (relative energy of TS is 3.2 kcal mol−1) as compared to that in the case of Me2(t-bipy). The energy required to preorganize the ligand for complexation is, therefore, lower in the case of Me2(t-phen) as compared to Me2(t-bipy), resulting in more favourable metal ion complexation in the case of Me2(t-phen) as compared to Me2(t-bipy). This is also reflected from the solvent extraction studies, where the distribution ratios of both Am3+ and Eu3+ were found to be higher in the case of Me2(t-phen) as compared to Me2(t-bipy) (Fig. 2).
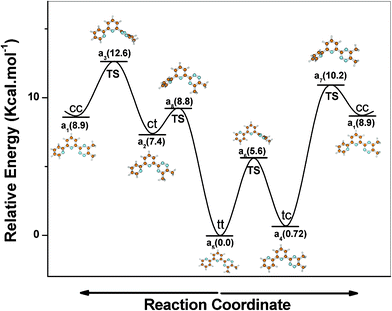 |
| | Fig. 4 The relative energies of different conformations of Me2(t-bipy) (minima and transition states) varying the angle of rotation around two rings with respect to the most stable ‘tt’ conformation | |
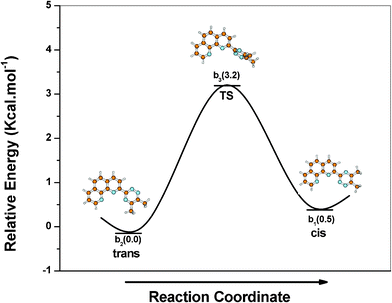 |
| | Fig. 5 The relative energies of different conformations of Me2(t-phen) (minima and transition states) varying the angle of rotation around two rings with respect to the most stable ‘trans’ conformation | |
Basicity of the ligands.
Protonation energies (Eprot) of the ligands were considered as a measurement of their basicity. The protonation energies were calculated from the difference in the energies of the most stable trans conformation of the ligand and its protonated form in cis-conformation as reported by Benay et al. in case of BTBPs.29 Protonation on the ‘N’ atom of the central pyridine ring was found to be most favourable in case of both the ligands and the protonation energy was found to be more negative in the case of t-phen (−263.2 kcal mol−1) as compared to that in the case of t-bipy (−254.9 kcal mol−1) resulting in higher basicity of t-phen. Benay et al. also, similarly, found a higher basicity of BT-phen (where 2,2′-bipyridine in BTBP was replaced by 1,10-phenanthroline) as compared to BTBP and they attributed this to the preorganized structure of BT-phen in the cis conformation.29
Analysis of frontier orbitals.
The energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were calculated for t-bipy and t-phen at the DFT level of theory. The HOMO–LUMO energy gap was found to be lower in the case of t-phen (92.1 kcal mol−1) as compared to t-bipy (98.3 kcal mol−1) (Fig. 6). The HOMO–LUMO gap is considered to be proportional to the absolute hardness.30,31 The lower HOMO–LUMO gap of t-phen, therefore, makes it softer than t-bipy. The higher selectivity of t-phen for Am3+ over Eu3+ as compared to t-bipy, as observed from the solvent extraction studies (Fig. 3), can be explained from the higher softness of t-phen. The higher selectivity of bis-1,2,4-triazinylpyridines (BTP) as compared to terpyridine (terpy) was similarly explained on the basis of lower energy gaps between the frontier orbitals in case of BTP.31 In the case of both the ligands, the triazine ring makes very little contribution in the π-donor HOMO and the major contributors here are the central and outer pyridine rings. The triazine ring, however, makes a significant contribution in the ligand′s LUMO, which can accept electrons from the metal centered occupied orbitals through back bonding.
 |
| | Fig. 6 A Walsh diagram showing the HOMO and LUMO and their energies (kcal mol−1) for Me2(t-bipy) and Me2(t-phen) | |
Computational studies on Am3+ and Eu3+ complexes of t-bipy and t-phen
Stronger ‘Am–N’ bonding was indicated by the higher sharing electron number (SEN) between the ‘Am’ and ‘N’ atoms in Am-complexes as compared to that between ‘Eu’ and ‘N’ atoms in Eu-complexes (Table 1). If we analyse the occupied valence orbitals of α spin in same cut off value, it is clear that the metal–ligand overlap is higher in the Am3+ complexes as compared to the Eu3+ complexes for both the ligands (Fig. 9 and ESI†). This indicates higher covalence in the Am3+ complexes as compared to the corresponding Eu3+ complexes resulting in selectivity of these classes of ligands for Am3+ over Eu3+. In the case of Am3+ complexes of the two ligands, t-bipy and t-phen, the HOMO of the complexes indicates that the overlap between the metal and ligand orbitals was higher in the t-phen complex and hence Am3+ complexation is further improved with t-phen as compared to t-bipy resulting in higher selectivity for Am3+ over Eu3+ using t-phen, and between the two ligands studied, t-phen shows higher covalence with the Am3+ ion. The triazine ring was reported to be a better electron acceptor as compared to the pyridine ring when their interaction with different trivalent actinide ions is considered.32 The major part of back bonding is, therefore, expected in the ‘M–N3’ bond because of the less negative charge on the ‘N3’ atom in the free ligands and significant contribution of the triazine ring in the LUMO of the ligands (Fig. 6). The shared electrons between the ‘M–N3’ bond, therefore, estimates the extent of metal–ligand bond strength or bond order in the complexes. For both the ligands, more electrons are shared between Am and N3 as compared to Eu and N3 atoms, resulting in stronger bonding in the Am-complexes, and hence selectivity of these ligands for Am3+ over Eu3+. A higher bond order or stronger metal–ligand bond in the Am-complex is also indicated by the shortening of the ‘Am–N3’ bond as compared to the ‘Eu-N3’ bond in the Eu-complexes (Table 1), in spite of marginally higher ionic radius of the Am3+ ion (1.230 Å) as compared to the Eu3+ ion (1.206 Å) for the same coordination number.33 If we compare the two ligands, t-bipy and t-phen, more electrons are shared between ‘M’ and ‘N3’ atoms in the case of t-phen for both the metal ions, indicating stronger bonding in the case of t-phen for both the metal ions. The difference in shared electrons in the ‘Am–N3’ bond and ‘Eu–N3’ bond is higher in the case of t-phen (0.082) as compared to the case of t-bipy (0.064), resulting in the higher selectivity of t-phen for Am3+ over Eu3+ as observed from the distribution studies. In the case of t-phen, all the three ‘M–N’ bonds are shorter in the Am-complex as compared to the Eu-complex, whereas in the case of t-bipy, the ‘M–N2’ bond is marginally longer in the Am-complex as compared to that in the Eu-complex. This also explains the higher selectivity of t-phen for Am3+ over Eu3+ as compared to t-bipy. Natural bond order (NBO) analysis or natural population analysis (NPA) is the technique where the orbitals used are the natural orbitals (eigenfunctions of the first order reduced density matrix) in spite of using the molecular orbitals directly for obtaining the occupancies and charges. This results in less basis set dependence as compared to other population analysis schemes, viz. Mulliken population analysis (MPA).34 The charges (Table 1) on the metal and three coordinating ‘N’ atoms in the Am3+ and Eu3+ complexes of t-bipy (Fig. 7(a)) and t-phen (Fig. 7(b)) were calculated using natural population analysis (NPA) as implemented in TURBOMOLE at their optimized geometries (Fig. 8). The more positive charge on Am as compared to Eu also supports higher metal to ligand back donation in the case of the Am-complex resulting in an increasing electronic charge on the coordinating nitrogen atoms in the Am-complex as compared to that in the Eu-complex.
 |
| | Fig. 7 The structures of the metal complexes of t-bipy (a) and t-phen (b) used for the DFT calculation (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H). H). | |
 |
| | Fig. 8 The optimized structures of Am3+ and Eu3+ complexes of t-bipy and t-phen. | |
 |
| | Fig. 9 The frontier orbitals of Am3+ and Eu3+ complexes of t-BTP and t-phen with same cut off value. | |
Table 1 The NPA charges on the directly bonded ‘N’ atoms and the metal ions (M), ‘M–N’ bond distances and shared electrons between these two bonded atoms in the Am3+ and Eu3+ complexes of t-bipy and t-phen (Fig. 8)
| |
NPA Charges |
Bond Length |
Shared Electrons |
| M |
N1 |
N2 |
N3 |
M–N1 |
M–N2 |
M–N3 |
M–N1 |
M–N2 |
M–N3 |
| t-bipy |
– |
−0.43 |
−0.41 |
−0.22 |
– |
– |
– |
– |
– |
– |
| Am-t-bipy |
2.09 |
−0.70 |
−0.64 |
−0.43 |
2.381 |
2.504 |
2.390 |
0.710 |
0.155 |
0.117 |
| Eu-t-bipy |
1.96 |
−0.69 |
−0.64 |
−0.39 |
2.456 |
2.500 |
2.509 |
0.706 |
0.034 |
0.053 |
| t-phen |
– |
−0.43 |
−0.40 |
−0.22 |
– |
– |
– |
– |
– |
– |
| Am-t-phen |
2.05 |
−0.69 |
−0.64 |
−0.44 |
2.469 |
2.483 |
2.401 |
0.470 |
0.254 |
0.175 |
| Eu-t-phen |
1.94 |
−0.67 |
−0.63 |
−0.42 |
2.515 |
2.505 |
2.480 |
0.532 |
0.160 |
0.093 |
Conclusions
A novel ligand, Me2(t-phen), with increased rigidity as compared to Me2(t-bipy), was synthesized and studied for the first time for the separation of trivalent actinides and lanthanides. The selectivity of these ‘N’ donor based ligands was attributed to the higher covalence in their Am-complexes as compared to their Eu-complexes, as indicated by the higher metal–ligand overlap in the case of Am3+-complexes as compared to the Eu3+-complexes. The stronger ‘Am–N’ bond as compared to the ‘Eu-N’ bond was also reflected in shorter ‘Am–N’ bonds in spite of the bigger size of the Am3+ ion, the higher number of shared electrons between ‘Am’ and ‘N’ atoms in the Am3+-complexes as compared to ‘Eu’ and ‘N’ atoms in the Eu3+-complexes and higher metal to ligand back donation in the Am-complex resulting in a higher residual positive charge on Am as compared to Eu. Improved extraction and separation of Am3+ over Eu3+ was experimentally observed using the ligand Me2(t-phen) as compared to Me2(t-bipy). The higher extraction of Am3+ with Me2(t-phen) was explained on the basis of its lower conformational energy requirement for complexation as compared to Me2t-bipy and the higher metal–ligand orbital overlap in the Am3+–t-phen complex as observed by analyzing the frontier orbitals of the Am3+ and Eu3+-complexes. The higher selectivity of t-phen for Am3+ over Eu3+ as compared to t-bipy was because of the higher softness of t-phen as compared to t-bipy as a result of the lower energy gap between the frontier orbitals and the higher value in the difference in shared electrons in the ‘Am–N3’ bond and ‘Eu-N3’ bond in case of t-phen as compared to that in case of t-bipy, resulting in a stronger Am3+-complex with respect to the Eu3+-complex in the case of t-phen as compared to that in the case of t-bipy.
Acknowledgements
DM and TKG would like to thank Dr S. K. Ghosh and Dr T. Mukherjee for their keen interest and constant encouragement.
References
- J.N. Mathur, M.S. Murali and K.L. Nash, Solvent Extr. Ion Exch., 2001, 19, 357 CrossRef CAS.
- C. Madic, M. J. Hudson, J. O. Liljenzin, J. P. Glatz, R. Nannicini, A. Facchini, Z. Kolarik and R. Odoj, Report EUR 19149EN, 2000 Search PubMed.
- Z. Kolarik, U. Müllich and F. Gassener, Solvent Extr. Ion Exch., 1999, 17, 23 CrossRef CAS.
- M.G.B. Drew, M.J. Hudson, P.B. Iveson, M.L. Russell, J.-O. Liljenzin, M. Skalberk, L. Spjuth and C. Madic, J. Chem. Soc., Dalton Trans., 1998, 2973 RSC.
- M.G.B. Drew, M.J. Hudson, P.B. Iveson, J.-O. Liljenzin, L. Spjuth, P.-Y. Cordier, A. Enarsson, C. Hill and C. Madic, J. Chem. Soc., Dalton Trans., 2000, 821 RSC.
- Z. Kolarik, U. Müllich and F. Gassener, Solvent Extr. Ion Exch., 1999, 17, 23 CrossRef CAS.
- M. J. Hudson, M. G. B. Drew, D. Guillaneux, M. L. Russell, P. B. Iveson and C. Madic, Inorg. Chem. Commun., 2001, 4, 12 CrossRef.
- M. J. Hudson, M. G. B. Drew, M. R. S. Foreman, C. Hill, N. Huet, C. Madic and T. G. A. Youngs, Dalton Trans., 2003, 1675 RSC.
- E. J. Corey, A. L. Borror and F. Thomas, J. Org. Chem., 1965, 30, 288 CrossRef CAS.
- F. H. Case, J. Heterocycl. Chem., 1970, 7, 1001 CrossRef CAS.
- T. Norby, A. Borje, L. Zhang and B. Akermark, Acta Chem. Scand., 1998, 52, 77 CrossRef.
-
P. K. Mohapatra, Ph.D. Thesis, University of Bombay, 1993. Search PubMed.
- L. Petit, C. Daul, C. Adamo and P. Maldivi, New J. Chem., 2007, 31, 1738 RSC.
- L. Petit, C. Adamo and P. Maldivi, Inorg. Chem., 2006, 45, 8517 CrossRef CAS.
- D. Guillaumont, THEOCHEM, 2006, 771, 105 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, K. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr., J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- MOLDEN visualization software is developed by CMBI, The Netherlands; http://www.cmbi.ru.nl/molden/molden.html.
- TURBOMOLE is program package developed by the Quantum Chemistry Group at the University of Karlsruhe, Germany, 1988: R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef.
- W. Kuechle, M. Dolg, H. Stoll and H. J. Preuss, Chem. Phys., 1994, 100, 7535 CAS.
- X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487 CrossRef CAS.
- X. Cao and M. Dolg, THEOCHEM, 2004, 673, 203 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730 CrossRef CAS.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348 CrossRef CAS.
- X. Cao and M. Dolg, THEOCHEM, 2002, 581, 139 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1993, 85, 441 CrossRef CAS.
- L. Oresmaa, M. Haukka, P. Vainiotalo and T.A. Pakkanen, J. Org. Chem., 2002, 67, 8216–8219 CrossRef CAS.
- G. Benay, R. Schurhammer and G. Wipff, Phys. Chem. Chem. Phys., 2011, 13, 2922 RSC.
- R.G. Pearson, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8440 CrossRef CAS.
- L. Petit, C. Daul, C. Adamo and P. Maldivi, New J. Chem., 2007, 31, 1738 RSC.
- P. Maldivi, P. Petit, C. Adamo and V. C. R. Vetere, Chemie, 2007, 10, 888 CAS.
- R.D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, A32, 751 CrossRef CAS.
-
D. Young, Computational Chemistry: A practical guide for applying techniques to real world problems, John Wiley & Sons, Inc.Canada, 2001, 100 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra00445c |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 




