Prediction of thermodynamic properties of adsorbed gases in zeolitic imidazolate frameworks†
Hedi
Amrouche
ab,
Benoit
Creton
a,
Flor
Siperstein
b and
Carlos
Nieto-Draghi
a
aIFP Energies nouvelles, 1 et 4, Avenue de Bois-Préau, 92852, Rueil-Malmaison, France. E-mail: carlos.nieto@ifpen.fr; Fax: +33 (0)1.47.52.70.25; Tel: +33(0)1.47.52.58.60
bSchool of Chemical Engineering and Analytical Science, The University of Manchester, P.O. Box 88, Sackville Street, Manchester, M60 1QD, U.K
First published on 13th April 2012
Abstract
In this work we propose an original methodology to predict the isosteric heat of adsorption of polar and non-polar gases adsorbed in different Zeolitic Imidazolate Framework (ZIF) materials, combining molecular simulation results with a quantitative structure–property relationship (QSPR) approach. The main contribution of our study is the development of a series of structural and molecular descriptors that are useful to describe the adsorption capability of adsorbents. A linear relationship is established to correlate the characteristics of gases and ZIF structures with the isosteric heat of adsorption. A simple tool to estimate the hydrophilic/hydrophobic nature of the solids studied is proposed based on the analysis of our simulation results. The promising approach shown in this work would be useful for the selection of organic linkers in the development of new hybrid organic–inorganic materials.
Introduction
Zeolitic Imidazolate Frameworks (ZIF) are promising porous materials for CO2 capture applications.1 These materials, which constitute a subfamily of Metal Organic Framework (MOF) materials, are generally built from a transition metal M, such as Zn2+, linked by functionalized imidazolate organic groups (RIm). Due to an Im–M–Im angle close to the 145° O–Si–O angle in zeolites, ZIF materials possess structures similar to zeolites.2The possibility of tuning the linker enables the creation of a huge variety of candidates for separation applications.3 Nevertheless, it is unreasonable to explore all linker–structure possible combinations, and efficient scanning methods are strongly desirable to guide the development of structures.4 Such methods must provide a direct connection between the topological and chemical nature of materials and the thermodynamic properties that describe the adsorption behaviour of gases. Molecular simulation appears to be an appropriate tool to scan MOF materials for CO2 capture.5 In our previous works,6,7 we highlighted interesting correlations between CO2 isosteric heat of adsorption and solid–gas interactions represented by organic linker dipole moment and van der Waals parameters. Predicting the performance of hypothetical ZIF/MOFs structures for separation applications would be considerably more efficient if general correlation principles were available. Considering the vast amount of information required in the development of correlative models, the use of highly efficient statistical approaches, such as Quantitative Structure–Property Relationship (QSPR), becomes evident.
Quantitative structure–property relationship
The most efficient way to deal with a large amount of information is to use correlative approaches, such as the QSPR, which are derived from the “Quantitative Structure–Activity Relationship” (QSAR) methods. These methods are largely used by the pharmaceutical research industry in Computer-Aided Drug Design (CADD), combined with molecular simulation methods at electronic, atomic and molecular levels. These approaches emerge as credible alternatives to reduce the costs of developing new chemicals while helping to reduce their impact on man and environment. Westmoreland et al.8 provide several examples of companies who have successfully used these techniques in different applications, obtaining real competitive advantages for some of them (e.g. DuPont, Dow Chemicals, etc.) through either patenting or developing new materials or products. These approaches have proven to be of capital interest in R&D activities due to their highly predictive nature in comparison with conventional macroscopic models, which generally have to be fitted on experimental data with the drawbacks that these acquisitions may entail.9 In a recent study on R&D activities in simulation-based engineering sponsored by the US National Science Foundation, one of the main findings was that the use of simulation techniques with correlative models leads to new and better products with important financial and time savings.10 For instance, Integrated Computational Materials Engineering (ICME) can lead to a reduction in the development time from 10–20 years to about 2–3 years with a return on investment ranging in between 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 9:1.10 The use of QSPR and other mathematical correlative methods in different industrial research areas from prediction of CO2 capture by solvents,11 jet fuel thermo-physical properties,12 and modelling catalytic activity of gases in zeolites is in continuous expansion.14 Despite the large amount of work devoted to the application of QSPR methods to predict thermo-physical properties of fluids,15 there are only a few examples devoted to the study of the adsorption phenomena of organic molecules in zeolites16 and activated carbon17 and only one has been applied to model hydrogen adsorption in MOFs.18
:1 to 9:1.10 The use of QSPR and other mathematical correlative methods in different industrial research areas from prediction of CO2 capture by solvents,11 jet fuel thermo-physical properties,12 and modelling catalytic activity of gases in zeolites is in continuous expansion.14 Despite the large amount of work devoted to the application of QSPR methods to predict thermo-physical properties of fluids,15 there are only a few examples devoted to the study of the adsorption phenomena of organic molecules in zeolites16 and activated carbon17 and only one has been applied to model hydrogen adsorption in MOFs.18
There is a complete lack of QSPR studies on adsorption in the literature aiming to correlate the behaviour of a wide range of gases in MOFs or ZIF. This lack of information may be due to (i) the difficulties to define or identify descriptors valid for solid materials and (ii) the lack of experimental data. In the following subsections we will try to contribute to bridging this gap by introducing a QSPR analysis defining and using several descriptors to represent the physical and chemical behaviour of adsorbed molecules in ZIF. To the best of our knowledge, this is the first work aiming at the development of solid descriptors able to characterise adsorptive properties integrating a molecular simulation-QSPR approach.
The QSPR methods enable correlating system's characteristics, which are represented by a series of descriptors, with a target property. In this work, we address the challenge of predicting the isosteric heat of adsorption qst°. The isosteric heat of adsorption as our output target has been chosen because it represents essential input data for the Pressure Swing Adsorption (PSA) process modelling.19 Additionally, Yazaydin et al. have recently shown that it represents a good descriptor for understanding the CO2 uptake in different MOF structures.5 Therefore we can identify two main types of descriptors, some associated with the solid and some associated with the adsorbed gases.
ZIF solids are constructed as a combination of molecular building blocks, i.e. organic linkers and metallic sites. In view of our previous results where we showed that metallic sites play a secondary role on adsorption (main adsorption sites are relatively far from Zn metals), we have decided to only define molecular descriptors for the organic linkers.7 In a first step, we identified about 60 possible descriptors containing information from the solids (lattice parameters, accessible surface areas, porosities, linker dipolar and quadrupolar moment, etc.) and the gases (atmospheric boiling temperature, molecular weight, dipolar and quadrupolar moments, etc.). We also considered the possibility of some mathematical operations over descriptor values (inverse, natural logarithm, exponential and cross products of descriptors). Additional descriptors for solid–gas interactions were also considered (a series of cross products between descriptors of both types). Only the most relevant descriptors were retained for building the predictive QSPR equations. For descriptors that are highly inter-correlated, i.e. hold essentially the same information, we only retained the ones that showed the highest correlation to the target thermodynamic property.
We selected the heat of adsorption at zero coverage to test the predictive capability of the QSPR approach over a database containing fourteen different Zn-based ZIF solids (see Table 1) composed from three different topologies and fourteen different functionalized imidazolate organic linkers (see Fig. 1) and eleven gas molecules (containing non-polar, quadrupolar and dipolar molecules) (see Table 2). A point to be considered is the permeability of the selected structures for all gases considered. The concept of pore window is somewhat artificial for judging the permeability of ZIF materials, due to the high flexibility of the organic linkers. In fact, ZIF-7 and Substituted Imidazolate Material (SIM-1)20 are solids with smaller window apertures than the ones reported in Table 1 (≤3 Å), however, both of these materials allow hydrocarbon molecules (C1 to C4) to diffuse inside their pores.21–24
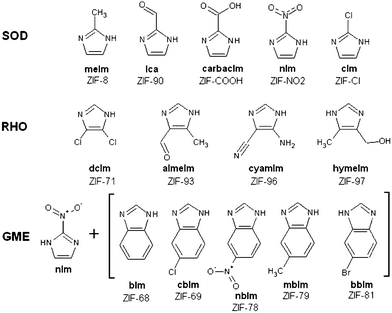 | ||
| Fig. 1 List of functionalized organic linkers of the different solids studied organized according to their topologies. The SOD and RHO functional groups are respectively localized in -2 and -4,-5 positions on the imidazolate linker. The GME structure is composed of two linkers: nitro-imidazolate, and a functionalized nitro benzo-imidazolium. The functional groups are localized on the -4 position of the benzoimidazole. | ||
| Name | Topology | Organic Linker | n fg | Cell volume/Å3 | d p [Å] | S a [Å2 g−1] | |μOL| [D] | |QOL| [D.Å] |
|---|---|---|---|---|---|---|---|---|
a
n
fg: number of functional group; dp: pore diameter calculated with the Material Studio package; Sa: accessible surface area calculated using the method developed by Düren et al.;13 |![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) OL| and |QOL| are, respectively, the dipole and quadrupole moment of the organic linkers. For information about the description of the SOD, RHO and GME topologies please see the web site http://izasc.ethz.ch/fmi/xsl/IZA-SC/ft.xsl.
b Hypothetical structures build from ZIF-8 using DFT computations.6 ZIF-NO2 differs from ZIF-65 by its transition metal (Zn rather Co). OL| and |QOL| are, respectively, the dipole and quadrupole moment of the organic linkers. For information about the description of the SOD, RHO and GME topologies please see the web site http://izasc.ethz.ch/fmi/xsl/IZA-SC/ft.xsl.
b Hypothetical structures build from ZIF-8 using DFT computations.6 ZIF-NO2 differs from ZIF-65 by its transition metal (Zn rather Co).
|
||||||||
| ZIF-8 | SOD | meIm | 1 | 4917.50 | 11.47 | 1395.14 | 1.18 | 8.97 |
| ZIF-90 | SOD | IcaIm | 1 | 5080.34 | 10.88 | 1269.42 | 3.15 | 13.46 |
| ZIF–COOHb | SOD | carboIm | 1 | 5131.07 | 10.99 | 1261.1 | 1.85 | 13.83 |
| ZIF–NO2b | SOD | nIm | 1 | 4980.17 | 10.61 | 949.78 | 3.33 | 12.65 |
| ZIF–Clb | SOD | cIm | 1 | 4950.98 | 11.10 | 1200.41 | 0.44 | 3.56 |
| ZIF-71 | RHO | dcIm | 2 | 23280.71 |
17.76 | 1137.2 | 0.52 | 10.30 |
| ZIF-93 | RHO | almeIm | 2 | 22801.20 |
17.04 | 968.13 | 5.66 | 25.54 |
| ZIF-96 | RHO | cyamIm | 2 | 22800.96 |
16.96 | 1251.7 | 5.20 | 28.02 |
| ZIF-97 | RHO | hymeIm | 2 | 22983.57 |
16.48 | 872.67 | 5.04 | 12.72 |
| ZIF-68 | GME | bIm | 1 | 22871.06 |
9.90 | 1060.83 | 6.63 | 16.23 |
| ZIF-69 | GME | cbIm | 1 | 22871.06 |
7.62 | 1003.1 | 7.59 | 17.05 |
| ZIF-78 | GME | nbIm | 1 | 22871.06 |
7.62 | 928.77 | 9.04 | 19.60 |
| ZIF-79 | GME | mbim | 1 | 22871.06 |
7.34 | 1012.54 | 8.05 | 10.49 |
| ZIF-81 | GME | bbIm | 1 | 22871.06 |
7.78 | 984.41 | 7.55 | 17.26 |
| Molecule name | Formula | Polar nature | T b[K] | Ref. |
|---|---|---|---|---|
| a np: non polar gas; Q: quadrupolar moment; μg: dipolar moment; D: Debye. Tb: Normal boiling temperature. | ||||
| Argon | Ar | np | 87.3 | 36 |
| Methane | CH4 | np | 111.6 | 37 |
| Ethane | C2H6 | np | 184.5 | 38 |
| Molecular nitrogen | N2 | Q (0.99 D.Å) | 77.9 | 39 |
| Molecular oxygen | O2 | Q (1.80 D.Å) | 90.0 | 40 |
| Carbon dioxide | CO2 | Q (5.06 D.Å) | 186.5 | 41 |
| Carbon monoxide | CO | μ g (0.03 D)/Q (0.06 D.Å) | 77.9 | 6 |
| Hydrogen sulphide | H2S | μ g (1.43 D)/Q (2.50 D.Å) | 211.0 | 42 |
| Sulphur dioxide | SO2 | μ g (1.63 D)/Q (7.19 D.Å) | 263.0 | 43 |
| Water | H2O | μ g (2.18 D)/Q (3.04 D.Å) | 363.0 | 44 |
| Acetonitrile | CH3CN | μ g (4.12 D)/Q (1.19 D.Å) | 383.0 | 45 |
Structural and gas adsorption studies on the selected solids have already been reported in different works. The GME structures have largely been explored in experimental25 and computational26–29 works. The RHO series have recently been published by Yaghi et al.30 Finally, the SOD series is composed from existing solids (ZIF-8,31 ZIF-9032) and hypothetical structures described in our previous work.6
Methods and simulation details
Thermodynamic properties were obtained using Grand Canonical Monte Carlo (GCMC) simulations with the GIBBS code.33 Solids and gases were considered as rigid and the solid–gas interactions were computed using a force field especially developed for ZIF structures.6 The assumption of rigid frameworks is justified because the calculations are performed at the limit of zero coverage, where structure deformation caused by the adsorbed gases is negligible. Table 2 indicates different force fields used to describe gas–gas interactions and to reproduce their different thermodynamic properties. To represent the electrostatic interactions and evaluate the multipole moments, partial charges have been computed using the Electrostatic Potential (ESP) methodology with Jaguar code:34 the B3LYP functional combined with the pseudo potential LanL2DZ was applied for the transition metal, and the double-ζ basis set 6-31G** was applied for the rest of the atoms. GME molecular structures were obtained from the Cambridge Crystallographic Database Centre (CCDC).35 RHO and SOD molecular structures were taken from the literature30 and our previous work,6 respectively. Isosteric heat of adsorption at zero coverage qst° was then computed at 303.15 K using (1).
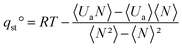
| (1) |
The choice of simulated results as input data was motivated by the lack of experimental data and to avoid possible inconsistencies generated by experimental uncertainties from different sources, from the quality of the sample, activation procedure, to uncertainties in the measurements and the extrapolation to zero coverage properties.
Simulated qst° results are compared to available experimental data to validate our approach (see Fig. 2). Experimental qst° values have been directly measured46 for CO2 and CH4 in ZIF-8 and ZIF-90, whereas values for GME structures have been inferred by using the van't Hoff equation over the experimental data reported in ref. 28. We observe a Mean Absolute Percentage Error (MAPE) of 12% between experimental data and GCMC simulations. The complete database, including our simulation results, experimental data and molecular descriptor values are available on simple request (technical details are reported in the Electronic Supplementary Information (ESI)†).
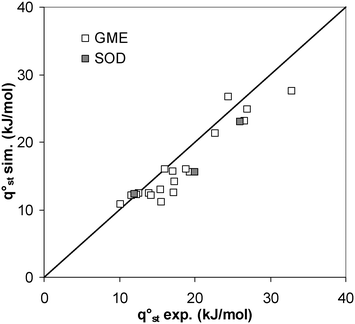 | ||
| Fig. 2 Comparison of qst° obtained with GCMC simulation with the available experimental data for SOD and GME topologies. | ||
Among the several statistical approaches available (Artificial Neural Network (ANN), Partial Least Squares (PLS), etc.), we selected the Genetic Function Approximation (GFA) as implemented in the Material Studio software.47 This choice was mainly motivated by the possibility to obtain a correlative equation composed from the most relevant descriptors. We choose the target predictive model to be multi-linear as described by eqn (2):
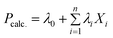 | (2) |
where Pcalc. is the target property, i runs over 1 to n, which corresponds to the maximum number of descriptors Xi admitted in the predictive equation and λi are weight factors. The GAF approach starts by establishing an initial population of equations, randomly chosen. The population of equations evolves through iterative operations: selection, crossover, and mutation of equation's terms (see Fig. S2 in ESI†). During the evolution process, the constructed equations are scored using the Friedman's Lack Of Fit (LOF) method instead of the well-known coefficient of determination R2, as the LOF favours small predictive equations.
The main descriptors used in the analysis, to represent the solid and gases are:
-Dipolar moment of organic linker μOL: There are two aspects to keep in mind to compute this descriptor: a) the linker definition and b) the way electrostatic interactions are determined.
a) Considering that gas adsorption in ZIF occurs exclusively in the surface of organic linkers7 a reasonable assumption is that thermodynamic properties will be governed by the chemical nature of the linkers. Therefore, we have introduced the idea that the polar character of the different ZIF can be represented by the linker dipolar moment (μOL) calculated from the partial charges used in the adsorption simulations (estimated with cluster calculations in Jaguar34).6 Two aspects influence the magnitude of dipolar moment of organic linkers: the electrostatic charges and the geometry of the linkers. We propose to use an isolated linker extracted from the cluster structure without the inclusion of Zn atoms, as shown in Scheme 1, where the organic linker can have a net charge, in a similar way to that reported in our previous work.6 The use of neutral linker (by taking the protonated version of the molecule) is not the most appropriate approach in our opinion, because the electronic environment of the linker would be significantly different from that encountered inside the solid structure.
![Linkers are extracted from the cluster (Zn[Linker]4) used to compute charge distribution without additional modification and its dipolar (quadrupolar) moment has been calculated using its point charge coordinates.](/image/article/2012/RA/c2ra00025c/c2ra00025c-s1.gif) | ||
| Scheme 1 Linkers are extracted from the cluster (Zn[Linker]4) used to compute charge distribution without additional modification and its dipolar (quadrupolar) moment has been calculated using its point charge coordinates. | ||
b) Many methods for estimating charge distributions can be used and the exact value of the linker dipole moment is strongly dependent on the method and geometry chosen to define the dipolar vector. Nevertheless, the relevant information to correlate the zero coverage heat of adsorption is the relative difference between dipolar moments on different linkers. A recent work of Watanabe et al. compares different ways of estimating point charges used in GCMC simulations.48 In addition, two different approaches based on the charge equilibration methods49 and group contribution method50 have been also successfully tested as an alternative to charge calculation using DFT. For the successful development of QSPRs the only constraint is that the dipolar moment of linkers should be determined in a similar way and in agreement with the charge distribution used for the adsorption simulations (in particular to estimate the different thermodynamic properties, such as isosteric heat of adsorption, Henry constant, adsorption isotherms, etc.).
GME structures have two different linkers, hence, they have two different dipolar moments. We decided to represent the GME structure by a unique dipolar moment, which is computed from the nIm and the RIm dipolar moments noted as μnIm and μRIm, respectively (see Fig. 1 for details). These values are weighted by the pore and channel accessible volume fraction, noted as ϑpore and ϑchannel, respectively. This choice is justified because RIm linkers are mainly distributed over the kno pores while the nIm are mostly placed on the gme and hpr pores, which are placed on the solid channels (pore volumes and GME structure description are reported in the ESI†). The GME dipole moment is defined in eqn (3).
| μGME = ϑporeμRIm + ϑchannelμnIm | (3) |
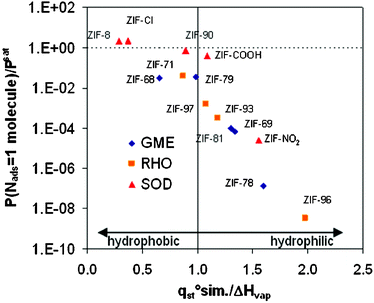
Taking into account that Vfree = Vpore + Vchannel, the ϑpore and ϑchannel accessible volume fractions are determined:
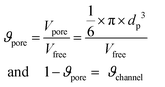 | (4) |
In this calculation we approximate pore volume as the volume of a sphere.
-Mean quadrupole moment of organic linkers |QOL|: these were computed as the norm of the diagonal elements of the quadrupolar matrix,
| |QOL| = (QXX2 + QYY2 + QZZ2)1/2 | (5) |
where QXX, QYY and QZZ are the diagonal elements of the matrix. This quantity is obtained similarly to the dipolar moment, using the point charge distributions and the organic linker geometry. Care must be taken to ensure that the matrix is diagonal, since the organic linkers have a net charge different from zero (due to the fact that the linker–metal ionic bond have been cut).
-Pore mean curvature H: this describes how a surface deviates from a flat plane. For real solid surfaces the calculation of this descriptor may become a difficult task, since it is obtained as 2H = ∇·![[n with combining circumflex]](https://www.rsc.org/images/entities/i_char_006e_0302.gif) , where is the unit normal vector of the surface.51 As a first approximation it is possible to consider solid pores as being simple spherical cavities. In this case the mean curvature H can be defined as 1/dp, where dp is the pore diameter (a flat surface has zero curvature). The Gaussian curvature K (defined as H2) has been used as a descriptor to take into account the dispersion contribution to the solid–gas energy terms for adsorption in zeolites.52,53 The curvature accounts, in an effective way for the degree of confinement of a fluid inside a pore, so it can be thought as a simple measure of the solid–fluid vdW interactions. The pore diameter was estimated as the diameter of the biggest sphere that fits inside the pores of the solid without touching the accessible surface area computed using the Düren et al.13 method. For GME structures, dp values were taken as the pore diameter of kno cages. Indeed, CO2 molecules are first adsorbed in the pores and then in the small channels.7 We have noticed that CO2 molecules adsorbed in small channels are forcibly aligned with the channel. Recently Babarao et al.54 suggested that the small channels of the GME solids are not accessible for CO2. The permeation of gases in ZIF structures is difficult to predict due to the high flexibility of the framework. We make the hypothesis that the adsorbed gases can easily access the main pores, while the permeation of small channels would depend on the size of the molecule and the applied pressure.6 In our opinion the debate about the permeation of gases in the channels of GME structures is still open; consequently, we decide to only consider the large pore diameter as the main quantity to estimate curvatures.
, where is the unit normal vector of the surface.51 As a first approximation it is possible to consider solid pores as being simple spherical cavities. In this case the mean curvature H can be defined as 1/dp, where dp is the pore diameter (a flat surface has zero curvature). The Gaussian curvature K (defined as H2) has been used as a descriptor to take into account the dispersion contribution to the solid–gas energy terms for adsorption in zeolites.52,53 The curvature accounts, in an effective way for the degree of confinement of a fluid inside a pore, so it can be thought as a simple measure of the solid–fluid vdW interactions. The pore diameter was estimated as the diameter of the biggest sphere that fits inside the pores of the solid without touching the accessible surface area computed using the Düren et al.13 method. For GME structures, dp values were taken as the pore diameter of kno cages. Indeed, CO2 molecules are first adsorbed in the pores and then in the small channels.7 We have noticed that CO2 molecules adsorbed in small channels are forcibly aligned with the channel. Recently Babarao et al.54 suggested that the small channels of the GME solids are not accessible for CO2. The permeation of gases in ZIF structures is difficult to predict due to the high flexibility of the framework. We make the hypothesis that the adsorbed gases can easily access the main pores, while the permeation of small channels would depend on the size of the molecule and the applied pressure.6 In our opinion the debate about the permeation of gases in the channels of GME structures is still open; consequently, we decide to only consider the large pore diameter as the main quantity to estimate curvatures.
-Number of functional groups nfg: this descriptor accounts for the number of non-aromatic functional groups that are present on each organic linker.
-Dipolar moment of adsorbed gas |μg|: this is the magnitude of the permanent dipolar moment of a molecule. This descriptor allows classification of the polar nature of the adsorbed gas.
-Atmospheric boiling temperature Tb of adsorbed gas: also known as the normal boiling temperature. We have chosen this descriptor for simplicity, since it is related to the heat of vaporization through the Clausius–Clapeyron equation, which is the amount of energy required to transfer a molecule embedded in a saturated liquid to the vapour phase. One may consider this magnitude as a crude estimation of the intrinsic cohesive energy between gas molecules. We have used Tb values reported in the literature computed with the different models of gases (see Table 2). The variation of the isosteric heat of adsorption obtained from GCMC simulations in function of the Tb of gases can be observed in Fig. 3. In general we observe a strong increment of qst° when Tb is increased. This effect becomes more important when the couple solid–gas is highly polar.
It is important to highlight that the thermodynamic properties of adsorbed gases used in our analysis were obtained from molecular simulations performed in the literature (with a MAPE < 2.9%). In cases where the required property was not available in the literature for a particular molecule, the appropriate simulation was performed in order to compute this quantity (see the ESI†).
 | ||
| Fig. 3 Variation of the isosteric heat of adsorption as function of the atmospheric boiling temperature (Tb) of all gases studied for the three topologies considered. | ||
Before carrying out calculations using the GFA approach, the total database was divided into two data subsets:
-The training set is intended to build the predictive models, and is chosen to be representative of the solids and gases studied. This represents 90% of the database;
-The test set constituted from the remaining solid/gas couples of the database. These latter are viewed as external values and used to test the predictive power of the equations and to select the best predictive model.
Most of the recent developments about correlations have been performed using QSPR approaches, which link input and output data through our statistical procedure, rather than physical phenomenological equations. Hence, it is not surprising that the interpretation of the correlative models obtained with QSPR methods usually lacks physical meaning. In fact the GFA procedure provides a series of multi-linear equations, more or less accurate, fitting the target property. In order to discriminate the most appropriate equation, we have constructed a consistency matrix relating the expected trends between the most relevant descriptors and the target property. In other words, we have introduced a form of physical consistency check to select the equation that preserves some physical intuition. Therefore a series of analysis was done, where qst° was studied as a function of variations of a single descriptor were carried out.
Results and discussion
The GFA procedure applied to the training set of our database leads to the following eqn (6):| qst° = (λ1|QOL| + λ2[nfg·H])s + (λ3ln(Tb))g + (λ4H·[|μOL|·|μg|])s/g | (6) |
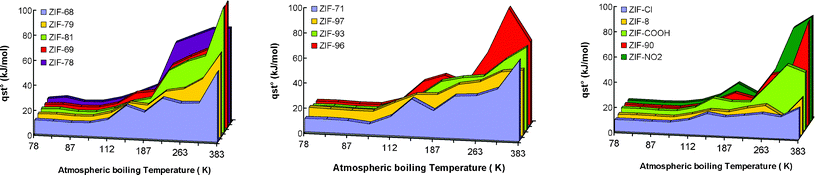
We can classify the selected descriptors according to three types of interactions, i.e. solid, gas and solid/gas (subindexes of eqn (6)). The different λi coefficients obtained for the correlation are reported in Table 3. The comparison between the simulation results and the QSPR correlation (6) for the isosteric heat of adsorption at zero coverage can be observed in Fig. 4. The accuracy of the QSPR model can be analyzed from the deviation obtained in reproducing the simulated qst° values. The Mean Absolute Error (MAE) and the MAPE are used to quantify the errors between predicted and simulated values. The lower MAE and MAPE, the better the prediction agree with experimental and/or simulation data.
 | ||
| Fig. 4 Comparison of the simulated and experimental isosteric heat of adsorption of different gases in different solids with prediction obtained using the QSPR model. Both sets of training and test are shown for consistency. | ||
| Coefficients | Units | |
|---|---|---|
| λ0 | −97.2651 | kJ mol−1 |
| λ1 | 0.52011 | kJ mol−1 D−1 Å−1 |
| λ2 | 95.8506 | kJ mol−1 Å |
| λ3 | 20.1537 | kJ mol−1 K−1 |
| λ4 | 9.6137 | kJ mol−1 D−2 Å |
The predicted results agree fairly well with simulated data for both the test and training sets (MAE about 5.7 kJ mol−1 and 5.6 kJ mol−1 and a MAPE about 24.5% and 23.8% for the test and the training sets). A detailed error analysis arranged by gas and solid topology is available in the supplementary information.†
It is worth spending some time analysing the form of eqn (6). Four descriptors of solids were considered: the polar nature of the solid (represented by |QOL| and |μOL|), the van der Waals interaction (represented by H and nfg). The adsorbed gas contributions are represented by |μg| and Tb, which are respectively a measure of its polar nature and its cohesive energy, i.e. the higher values of |μg| or Tb indicate stronger gas–gas interactions.
Eqn (6) agrees with some physical intuitions.55 Indeed any increment on solid and/or gas descriptors results in higher values for qst°. For example, the higher the multipole moments, the stronger will be the electrostatic interactions. Nevertheless, eqn (6) takes into account that for non-polar gases, such as methane, there is no contribution from the linker dipole moment |μOL| to qst°. For polar gases, higher values of |μOL| will lead to higher values of qst°. Moreover, the analysis reveals that the degree of polarity of the gas is also important. For example, gases having quadrupolar moment (such as CO2) are less influenced by solids having highly polar linkers than polar gases (like SO2).
The higher the gas polarity, the higher the increase of qst° will be when we increase |μOL|.
The dispersive–repulsive terms represented by H and nfg take into account the solid topology and the chemical nature of the organic linkers implicitly. Eqn (6) shows that qst° increases with H, if we consider solids with a single functional group. Nevertheless, it is important to mention that solids with RHO topologies have bi-substituted organic linkers, while SOD and GME possess linkers with only one non-aromatic functional group. Therefore, if we consider the product (nfg·H) the vdW contributions are grouped by topology, the smaller values are obtained for the SOD, followed by the RHO and finally by the GME structures.
It is not straightforward to discriminate which kind of fluid (non polar, quadrupolar or polar) is better (or worse) described by the model. It seems that there is no particular correlation between polar nature of the gases and the obtained deviation. We can notice however that the lowest MAPE deviations are for CO2, SO2, CH4 and the highest ones for H2O and N2. We can observe the following order from lowest to highest MAPE,
| CO2 < SO2 < CH4 < CH3CN < C2H6 < O2 < H2S < Ar < CO <H2O < N2 |
Our analysis reveals that the QSPR model for qst° is slightly more accurate for the family of solids with GME topology (17.8%), followed by the RHO (23.5%) and SOD (33.1%) topologies, respectively. The fluid responsible for the higher deviation observed in the SOD and GME topologies is water. In fact, our model fails to describe the strong hydrophobic character of ZIF-8, ZIF-Cl and ZIF-68 by predicting too high values of qst° compared with simulation results. In some cases water can form Hydrogen Bonds (HB) with some functional groups of the solids, which can be considered as highly hydrophilic. In other cases water forms strong self-association (by HB clustering) when adsorbed over solids having unfavourable interactions, so the solid can be considered as hydrophobic. The lack of information about the behaviour of water in MOF is a known problem.4 Consequently, we have decided to analyse the behaviour of water obtained in our results. In view of the poor description of our model to reproduce solid–water interactions (we observe a MAPE of 34%) we concentrate the analysis on our results obtained with GCMC simulations.
The behaviour of water adsorbed in hydrophobic solids, such as dealuminated zeolites and clays have been extensively studied by different works.56,57 In the case of hydrophobic silicalite zeolites it was found that water behaves as a nanodroplet, trying to close its hydrogen-bond network onto itself with a few short-lived dangling OH groups, while water in hydrophilic zeolites “opens up” to form weak hydrogen bonds with the zeolite oxygen atoms and strong interactions with the extra-framework ions.58 Similar conclusion have been obtained for the adsorption of water in ion-exchanged rho-MOF (ZIF charged structures having extraframework ions).59 In order to understand the process of intrusion/extrusion of water, Caillez et al. have introduced a potential energy balance for water adsorbed in hydrophobic zeolites with respect to bulk water.60
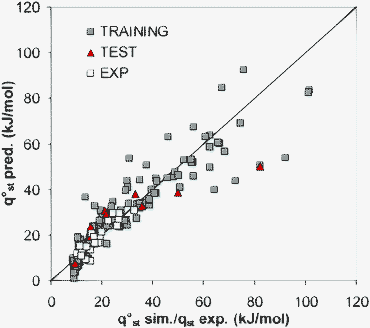
A detailed structural and thermodynamical analysis of water molecules in hydrophobic and hydrophilic ZIF is out of the scope of this work. However, we can exploit our results in order to estimate the hydrophilic or hydrophobic nature of the surface of a ZIF structure by comparing the solid-water interactions with respect to water–water interactions in bulk. We can define two quantities: (i) the ratio [qst°/ΔHvap]water, which is an estimator of the relative affinity of a water molecule to approach a solid surface embedded in liquid water, and (ii) the ratio [P(Nads = 1 molecule)/Psat]water gives an idea of how far water adsorbed at Henry's regime is with respect to saturated water at the same temperature. Here Psat and P(Nads = 1) are the saturation pressure and the pressure at which 1 molecule of water is adsorbed per unit cell, respectively. The relation between the two aforementioned quantities can be observed in Fig. 5 for different ZIF studied.
 | ||
| Fig. 5 Comparison of the hydrophobic/hydrophilic nature of the different solids studied. In this figure we have used ΔHvap = 46.5572 kJ mol−1 and Psat = 0.050067 bar obtained with the TIP4P model of water at 303 K.61,62 | ||
According to Fig. 5, ZIF-8 and ZIF-Cl are highly hydrophobic, followed by ZIF-68, ZIF-71, ZIF-90 and ZIF-79. The remaining solids are hydrophilic to different extents. The trend observed confirms the expected hydrophobic nature of solids that have unfavourable functional groups for water adsorption (–[CH3], –[Cl] and pure aromatic rings). The opposite trend is also confirmed, as most favourable functional groups (–[CHO:CH3], –[NO2], –[CN:NH2]) for water lead to the most hydrophilic solids, i.e. ZIF-96, ZIF-NO2 and ZIF-78. In hydrophilic solids water molecules at low coverage are probably stabilised by the formation of strong solid–water HB or by dipole-like interactions. The particular case of ZIF-69 and ZIF-81 with –[Cl] and –[Br] functional groups, respectively, is interesting. Both solids have hydrophobic linkers and, nevertheless exhibit a slightly hydrophilic character. These materials have a GME structure, which possesses two functional groups: nIm, which is a strong hydrophilic linker and substituted RIm linker (see Fig. 1). Consequently, the hydrophilic nature of GME structures depends on a complicated balance of interactions between both types of linkers. The hydrophobic nature of ZIF-71 and ZIF-8 has been confirmed by means of molecular simulations63 and experimental measurements,64 respectively. Unfortunately there are no additional experimental results in the literature concerning water adsorption in ZIF to confirm the behaviour proposed in Fig. 5.
Conclusions
The objective of this work is to open an unexplored way for the prediction and comprehension of adsorption phenomena in ZIF materials. We proposed an equation able to predict the isosteric heat of adsorption for ZIF materials from four descriptors. The predictive power of this equation is satisfactory, even if the precision could be improved. Nevertheless, the principal purpose of this work was to evaluate the potential of a QSPR approach to predict thermodynamics properties of ZIF structures. The agreement obtained with simulated data encourages the development of new models based on the same approach. The accuracy of our method could be improved: (i) by extending the size of the database used, (ii) by including more ZIF or MOF structures, (iii) by the development of additional descriptors for solid or gas, (iv) by using approaches leading to non-linear models such as the well known Artificial Neural Networks (see the model proposed in the ESI†).We propose, in addition, a simple tool to estimate the hydrophobic/hydrophilic nature of ZIF by using two quantities based on thermodynamic data of water and solids.
The development of this kind of tool would constitute powerful way to summarize results obtained for the different solids. This approach helps the comprehension of adsorption phenomena by proposing the most influent factors. The properties of hypothetical solids would be predicted before their synthesis. This approach would hence orientate the development of ZIF materials as a function of the application desired, with reasonable computing cost.
Acknowledgements
We would like to thanks Mr. E. Garcia, Dr J. Pérez-Pellitero, Dr G. Pirngruber and Dr Caroline Mellot-Draznieks for fruitful discussions. All DFT optimisation and GCMC calculations have been performed at IFPEN HPC centre and at IDRIS/CINES HPC centres within the projects x2010086335 funded by GENCI.References
- A. Phan, C .J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keefe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS.
- J-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H. K. Jeong, P. B. Balbuena and H-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791 CrossRef CAS.
- A. Ö Yazaydın, R. Q. Snurr, T-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198 CrossRef.
- H. Amrouche, S. Aguado, J. Pérez-Pellitero, C. Chizallet, F. Siperstein, D. Farrusseng, N. Bats and C. Nieto-Draghi, J. Phys. Chem. C, 2011, 115, 16425 CAS.
- J. Pérez-Pellitero, H. Amrouche, F. Siperstein, G. Pirngruber, C. Nieto-Draghi, G. Chaplais, A. Simon-Masseron, D. Bazer-Bachi, D. Peralta and N. Bats, Chem.–Eur. J., 2010, 16, 1560 CrossRef.
- P. Westmoreland, P. Kollman, A. Chaka, P. Cummings, K. Morokuma, M. Neurock, E. Stechel, P. Vashishta, Applications of Molecular and Materials Modeling, Report of the World Technology Evaluation Center (WTEC), International Technology Research Institute, 2002, www.wtec.org/pdf/molmodel.pdf Search PubMed.
- P. Ungerer, B. Tavitian, A. Boutin, F. Montel, Applications of molecular simulation in the oil and gas industry. Editions Technip, 2005 Search PubMed.
- S. C. Glotzer, S. Kim, P. T. Cummings, A. Deshmukh, M. Head-Gordon, G. Karniadakis, L. Petzold, C. Sagui, M. Shinozuka, International Assesment of research and development in simulation-based engineering and science, Report of the World Technology Evaluation Center (WTEC). International Technology Research Institute, 2009, www.wtec.org/sbes/SBES-GlobalFinalReport.pdf Search PubMed.
- F. Porcheron, A. Gilbert, P. Mougin and A. Wender, Environ. Sci. Technol., 2011, 45, 2486 CrossRef CAS.
- D. A. Saldana, L. Starck, P. Mougin, B. Rousseau, L. Pidol, N. Jeuland and B. Creton, Energy Fuels, 2011, 25, 3900 CAS.
- T. Düren, F. Millange, G. Férey, K. S. Walton and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 15350 Search PubMed.
- P. Leflaive, G. Pirngruber, A. Faraj, P. Martin, G. V. Baron and J. F. M. Denayer, Microporous Mesoporous Mater., 2010, 132, 246 CrossRef CAS.
- A. R. Katritzky, M. Kuanar, S. Slavov and C. D. Hall, Chem. Rev., 2010, 110, 5714 CrossRef CAS.
- A. Goulon, A. Faraj, G. Pirngruber, M. Jacquin, F. Porcheron, P. Leflaive, P. Martin, G. V. Baron and J. F. M. Denayer, Catal. Today, 2011, 159, 74 CrossRef CAS.
- B. Lei, Y. Ma, J. Li, H. Liu, X. Yao and P. Gramatica, Atmos. Environ., 2010, 44, 2954 CrossRef CAS.
- D. Kim, J. Kim, D. H. Jung, T. B. Lee, S. B. Choi, J. H. Yoon, J. Kim, K. Choi and S.-H. Choi, Catal. Today, 2007, 120, 317 CrossRef CAS.
- D. P. Valezuela, A. L. Myers, Adsorption Equilibrium Data Handbook, Prentice Hall, Englewood Cliffs, USA, 1989 Search PubMed.
- D. Farrusseng, S. Aguado, J. Canivet, FR09/04488 Patent R09/04488, 2009 Search PubMed.
- S. Aguado, G. Bergeret, M. P. Titus, V. Moizan, C. Nieto-Draghi, N. Bats and D. Farrusseng, New J. Chem., 2011, 35, 546 RSC.
- C. Gücüyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704 CrossRef.
- S. Aguado, C. H. Nicolas, V. Moizan-Baslé, C. Nieto, H. Amrouche, N. Bats, N. Audebrand and D. Farrusseng, New J. Chem., 2011, 35, 41 RSC.
- S. C. Reyes, J. G. Santiesteban, Z. Ni, P. Kortunov, J. Zengel, H. W. Deckman, US 2009/0211440 A1 US Patent, 2009 Search PubMed.
- R. B. Rankin, J. Liu, A. D. Kulkarni and J. K. Johnson, J. Phys. Chem. C, 2009, 113(39), 16906 CAS.
- R. Babarao, S. Dai and D-e. Jiang, J. Phys. Chem. C, 2011, 115, 8126–8135 CAS.
- J. Liu, S. Keskin, D. S. Sholl and J. Karl Johnson, J. Phys. Chem. C, 2011, 115, 12560 CAS.
- R. Banerjee, H. Furukawa, D. Britt, M. Knobler O'Keeffe and O. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS.
- Y. Liu, H. L. Liu, Y. Hu and J. W. Jiang, J. Phys. Chem. B, 2010, 114, 2820 CrossRef CAS.
- W. Morris, B. Leung, H. Furukawa, O. K. Yaghi, N. He, H. Hayashi, Y. Houndonougbo, M. Asta, B. B. Laird and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 11006 CrossRef CAS.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557 CrossRef CAS.
- W. Morris, C. J. Doonan, H. Furukawa, R. Banerjee and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 12626 CrossRef CAS.
- P. Ungerer, C. Nieto-Draghi, V. Lachet, A. Wender, A. Di Lella, A. Boutin, B. Rousseau and A. Fuchs, Mol. Simul., 2007, 33, 287 CrossRef CAS.
- Jaguar version 7.0, Schrödinger, LLC., New York, NY, 2007.
- The Cambridge Crystallographic Data Centre, www.ccdc.cam.ac.uk.
- J. Vrabec, J. Stoll and H. Hasse, J. Phys. Chem. B, 2001, 105, 12126 CrossRef CAS.
- D. Moller, J. Oprzynski, A. Muller and J. Fischer, Mol. Phys., 1992, 75, 363 CrossRef.
- P. Ungerer, C. Beauvais, J. Delhommelle, A. Boutin, B. Rousseau and A. Fuchs, J. Chem. Phys., 2000, 112, 5499 CrossRef CAS.
- J. Delhommelle, PhD Thesis, Universite de Paris XI, Orsay, France, 2000 Search PubMed.
- Y. Boutard, P. Ungerer, J. M. Teuler, M. G. Ahunbay, S. F. Sabater, J. Perez-Pellitero, A. D. Mackie and E. Bourasseau, Fluid Phase Equilib., 2005, 236, 25 CrossRef CAS.
- G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021 CrossRef.
- T. Kristóf and J. Liszi, J. Phys. Chem. B, 1997, 101, 5480 CrossRef.
- E. El Ahmar, B. Creton, A. Valtz, C. Coquelet, V. Lachet, D. Richon and P. Ungerer, Fluid Phase Equilib., 2011, 304, 21 CrossRef CAS.
- W. L. Jorgensen, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- D. M. F. Edwards, P. A. Madden and I. R. McDonald, Mol. Phys., 1984, 51, 1141 CrossRef CAS.
- S. Aguado, J. Canivet, M. Pera-Titus, H. Amrouche, J. Perez-Pellitero3, V. Moizan-Basle, C. Nieto-Draghi and D. Farrusseng, submitted Search PubMed.
- Materials Studio, Release 5.0, Accelrys Software Inc., San Diego, 2009 Search PubMed.
- T. Watanabe, T. A. Manz and D. S. Sholl, J. Phys. Chem. C, 2011, 115, 4824 CAS.
- C. E. Wilmer and R. Q. Snurr, Chem. Eng. J., 2010, 171, 775 CrossRef.
- Q. Xu and C. Zhong, J. Phys. Chem. C, 2010, 114, 5035 CAS.
- K. Miyasaka and O. Terasaki, Angew. Chem., Int. Ed., 2010, 49, 8867 CrossRef CAS.
- Z. Blum, S. T. Hyde and B. W. Ninham, J. Phys. Chem., 1993, 97, 661 CrossRef CAS.
- R. Thomasson, S. Lidin and S. Anderson, Angew. Chem., Int. Ed. Engl., 1987, 26, 1017 CrossRef.
- R. Babarao, S. Dai and D-E. Jiang, J. Phys. Chem. C, 2011, 115, 8126 CAS.
- R. T. Yang, Adsorbents: Fundamental and Applications, Wiley, New Jersey, 1989 Search PubMed.
- B. Bougeard and K. Smirnov, Phys. Chem. Chem. Phys., 2007, 9, 226 RSC.
- N. Desbiens, I. Demachy, A. H. Fuchs, H. Kirsch-Rodeschini, M. Soulard and J. Patarin, Angew. Chem., Int. Ed., 2005, 44, 5310–5313 CrossRef CAS.
- F. X. Coudert, F. Cailliez, R. Vuilleumier, A. H. Fuchs and A. Boutin, Faraday Discuss., 2009, 141, 377 RSC.
- A. Nalaparaju, R. Babarao, X. S. Zhao and J. W. Jiang, ACS Nano, 2009, 9, 2563 CrossRef.
- F. Cailliez, M. Trzpit, M. Soulard, I. Demachy, A. Boutin, J. Patarin and A. H. Fuchs, Phys. Chem. Chem. Phys., 2008, 10, 4817 RSC.
- M. Lísal, W. R. Smith and I. Nezbeda, Fluid Phase Equilib., 2001, 181, 127 CrossRef.
- C. Vega, J. L. F. Abascal and I. Nezbeda, J. Chem. Phys., 2006, 125, 034503 CrossRef CAS.
- A. Nalaparaju, X. S. Zhao and J. W. Jiang, J. Phys. Chem. C, 2010, 114, 11542 CAS.
- P. Küsgens, M. Rose, I. Senkovsk, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available: Simulation details as well as complementary information about the QSPR method and the full solid–gas property database is available in the ESI. See DOI: 10.1039/c2ra00025c/ |
| This journal is © The Royal Society of Chemistry 2012 |