Platinum(II) and palladium(II) complexes derived from 1-ferrocenylmethyl-3,5-diphenylpyrazole. Coordination, cyclometallation or transannulation?†
Concepción
López
*a,
Asensio
González
b,
Ramon
Bosque
a,
Pradipta K.
Basu
ab,
Mercè
Font-Bardía
c and
Teresa
Calvet
d
aDepartament de Química Inorgànica, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1-11. E-08028-Barcelona, SPAIN
bLaboratori de Química Orgánica, Facultat de Farmacia, Universitat de Barcelona, Pl. Pius XII s/n. E-08028-Barcelona, SPAIN
cUnitat de Difracció de Raigs-X. Centre Científic i Tecnològic de la Universitat de Barcelona. Universitat de Barcelona, Solé i Sabaris 1-3, 08028-Barcelona, SPAIN
dDepartament de Cristal·lografia Mineralogía i Dipòsits Minerals. Facultat de Geologia. Universitat de Barcelona, Martí i Franquès s/n. E-08028-Barcelona, SPAIN
First published on 10th January 2012
Abstract
The synthesis and characterization of the novel pyrazole derivative [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2) {Fc = (η5–C5H5)Fe(η5–C5H4)–} with a ferrocenylmethyl substituent on position 1 of the heterocycle is described. The study of the reactivity of 2 with cis-[MCl2L2] (M = Pt and L = dmso or M = Pd and L = dmso or CH3CN), Pd(AcO)2 or Na2[PdCl4] under different experimental conditions, has allowed us to isolate and characterize a wide variety of platinum(II) or palladium(II) complexes: trans-[Pt{1-(Fc–CH2)-3,5-Ph2-(C3HN2)}Cl2(dmso)] (3), the cis- isomers of [M{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] {M = Pt (4) or Pd (7)}, trans-[Pd{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}2Cl2] (8), the cyclometallated compounds [M{1-(Fc–CH2)-(3–C6H4)-5-Ph–(C3HN2)}Cl(L)] {with M = Pt and L = dmso (5) or PPh3 (6) or M = Pd and L = PPh3 (9)} and the palladium(II) complex [Pd{1-[(η5–C5H4)Fe{(η5–C5H4)–CH2]-3,5-Ph2–(C3HN2)}Cl(PPh3)] (10) that arises from a transannulation process. The crystal structures of the free ligand 2 and compounds 4, 7, 9 and 10 are also reported and confirm the cis- disposition of the Cl− ligands in 4 and 7, the trans- arrangement of the phosphorous and the nitrogen atoms in 9 and 10, the mode of binding of the ligand in 4, 7, 9 and 10 and the nature of the metallated carbon atom {C(sp2, phenyl) in 9 or the C(sp2, ferrocenyl) of the C5H5 ring in 10}. In order to rationalize the different nature of the products isolated in the reactions of 2 with Pd(AcO)2 or Na2[PdCl4] and NaAcO density functional theory (DFT) calculations of the complexes have also been carried out.
Introduction
Ferrocene heterocyclic systems have attracted great interest in recent years 1–3 because of their physical and chemical properties as well as their applications in a wide variety of areas including homogeneous catalysis1,2d,3c and biomedicine.2e In this class of compounds, the presence of heterocycles with one or more atoms with good donor abilities is especially interesting in view of their use as ligands for transition metals to give heteropolymetallic complexes in which the existence of two or more proximal metal ions may induce a co-operative effect.1–4On the other hand, the investigation of palladium(II) and platinum(II) coordination complexes with pyrazoles as ligands is one of the research areas that has undergone rapid development during recent years.5–8 Compounds of this kind exhibit greater antitumoral activity and lower toxicity than cis-[PtCl2(NH3)2] or antibacterial activity have been reported.6c Furthermore, some examples of their utility in macromolecular chemistry,7a or in homogeneous catalysis7b have also been published.
In addition, cyclometallated Pd(II) and Pt(II) complexes of N– donor ligands are particularly relevant due to their photophysical properties, biological activity and applications in homogeneous catalysis, or as building blocs in supra- and macromolecular chemistry.9–15
Despite of: a) the increasing interest in new ferrocene derivatives containing pyrazole units,16 b) their use as ligands in front of transition metals,4b,4c,7 c) the study of heterodi-, tri- or in general polymetallic complexes with ferrocenyl units,1,3,4 or d) the relevance of palladium(II) and platinum(II) in synthesis,9,10a,b,14 only a few palladium(II) complexes containing hybrid ferrocene-pyrazole units have been reported so far,4b,4c and as far as we know, platinum(II) derivatives are unknown.
We have recently described the syntheses of [3,5-Ph2-4-(Fc–CH2)–(C3HN2)] (1) {Fc = (η5–C5H5)Fe(η5–C5H4)–} (Fig. 1) and a few palladium(II) and platinum(II) complexes where 1 behaves as an (N) or [C(phenyl), N(pyrazol)]− ligand.4b In view of the results obtained in these studies, and in order to elucidate whether the position of the Fc–CH2– unit on the heterocycle could affect: a) the reactivity of this sort of compound, b) the nature of the palladium(II) and platinum(II) complexes formed or c) their properties, we decided to prepare and characterize the new ligand [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2) (Fig. 1).
![Chemical formulae of 1-methyl-3,5-diphenyl-4-ferrocenylene-pyrazole (1) studied before4c and of the new ligand [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2) prepared and used in this work.](/image/article/2012/RA/c1ra01080h/c1ra01080h-f1.gif) | ||
| Fig. 1 Chemical formulae of 1-methyl-3,5-diphenyl-4-ferrocenylene-pyrazole (1) studied before4c and of the new ligand [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2) prepared and used in this work. | ||
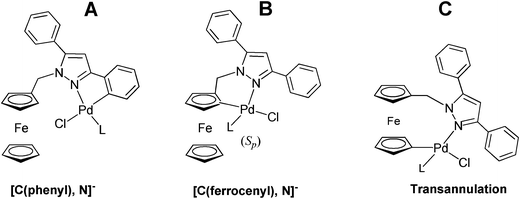
In compound 2, the presence of the Fc–CH2– unit on position 1 of the heterocycle has an additional interest due to the existence of different types of carbon atoms [C(phenyl) and C(ferrocenyl)] susceptible to metallation. The activation of the σ[C(phenyl)–H] bond would produce five-membered metallacycles (Fig. 2, A); while the metallation of the C5H4 ring of the ferrocenyl unit, that induces planar chirality, would give chiral compounds (Rp or Sp isomers) containing six-membered rings (Fig. 2, B). Moreover, since a few uncommon examples of palladacycles arising from a transannulation process have been recently reported,4a,18 the formation of palladium(II) or platinum(II) complexes arising from the activation of the σ[C(ferrocene)–H] bond of the unsubstituted C5H5 ring of the Fc moiety (Fig. 2, C) or complexes containing [C,N,C']2− pincer ligands (Fig. 2, D and E) formed by the activation of two σ(C−H) bonds cannot be ruled out.19
![Schematic view of the different types of pallada- and platinacycles that could be formed through: a) activation of a σ(Cphenyl–H) bond (A) or one of the two σ[C(ferrocenyl)–H] bonds on the ortho site of the C5H4 ring (B) {in this case two enantiomers: (Sp) and (Rp) could be formed}, b) from a transannulation process (C) or c) from a more complex reaction involving a double cyclometallation (D) or a transannulation and a cyclometallation reaction simultaneously (E).](/image/article/2012/RA/c1ra01080h/c1ra01080h-f2.gif) | ||
| Fig. 2 Schematic view of the different types of pallada- and platinacycles that could be formed through: a) activation of a σ(Cphenyl–H) bond (A) or one of the two σ[C(ferrocenyl)–H] bonds on the ortho site of the C5H4 ring (B) {in this case two enantiomers: (Sp) and (Rp) could be formed}, b) from a transannulation process (C) or c) from a more complex reaction involving a double cyclometallation (D) or a transannulation and a cyclometallation reaction simultaneously (E). | ||
In this paper we present the synthesis of ligand 2 and the study of its coordination chemistry in front of palladium(II) and platinum(II) in view of the potential applications of the complexes in different fields.
Results and discussion
The ligand
The new ferrocene derivative [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2) was prepared in good yield (96%) by electrophillic coupling at the nitrogen of 3,5-diphenylpyrazole using ferrocenylmethanol in a two-phase dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aqueous tetrafluoroboric acid (Scheme 1).20–21
:aqueous tetrafluoroboric acid (Scheme 1).20–21
 | ||
| Scheme 1 i) in CH2Cl2, treatment with aqueous HBF4, 1 h at room temperature. | ||
Elemental analyses (experimental section) of 2 were consistent with the proposed formula, its mass spectra showed a peak at m/z = 419.1, that agrees with the value expected for the {[M] + H}+ cation and the infrared spectrum of 2 exhibited the typical bands due to monosubstituted ferrocene derivatives.22
The X-ray crystal structure of 2 is depicted in Fig. 3. Average values of bond lengths [Fe–Cring: 2.07(4) Å and Cring–Cring: 1.43(3) Å] and angles of the Fc unit fall in the range reported for most monosubstituted ferrocene derivatives.23 The two pentagonal rings are planar, nearly parallel (tilt angle = 1.69°) and they deviate by ca. 18° from the ideal eclipsed conformation.
![ORTEP plot of [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2), 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity. Selected bond lengths (in Å) and angles (in deg): C(10)–C(11), 1.513(5); C(11)–N(2), 1.449(4); N(1)–C(12), 1.318(4); C(12)–C(13), 1.390(5); C(13)–C(14), 1.393(4); C(14)–N(2), 1.340(4); N(1)–N(2), 1.367(4); C(14)–C(15), 1.514(4); 1.480(4); C(14)–C(21), 1.480(4); Fe–C (average value), 2.07(4) (C–C) of Fc (average value), 1.43(3); C(6)–C(10)–C(9), 105.4(3); N(2)–C(11)–C(10), 111.2(3), C(6)–C(10)–C(11), 125.0(3); C(9)–C(10)–C(11), 129.3(3); N(1)–C(12)–C(13), 109.2(3); N(1)–C(12)–C(15), 120.8(3); N(2)–C(14)–C(13), 103.7(8); N(2)–C(14)–C(11), 123.3(3); C(12)–C(13)–C(14), 108.1(3) and C(13)–C(14)–C(21), 133.0(3).](/image/article/2012/RA/c1ra01080h/c1ra01080h-f3.gif) | ||
| Fig. 3 ORTEP plot of [1-(Fc–CH2)-3,5-Ph2–(C3HN2)] (2), 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity. Selected bond lengths (in Å) and angles (in deg): C(10)–C(11), 1.513(5); C(11)–N(2), 1.449(4); N(1)–C(12), 1.318(4); C(12)–C(13), 1.390(5); C(13)–C(14), 1.393(4); C(14)–N(2), 1.340(4); N(1)–N(2), 1.367(4); C(14)–C(15), 1.514(4); 1.480(4); C(14)–C(21), 1.480(4); Fe–C (average value), 2.07(4) (C–C) of Fc (average value), 1.43(3); C(6)–C(10)–C(9), 105.4(3); N(2)–C(11)–C(10), 111.2(3), C(6)–C(10)–C(11), 125.0(3); C(9)–C(10)–C(11), 129.3(3); N(1)–C(12)–C(13), 109.2(3); N(1)–C(12)–C(15), 120.8(3); N(2)–C(14)–C(13), 103.7(8); N(2)–C(14)–C(11), 123.3(3); C(12)–C(13)–C(14), 108.1(3) and C(13)–C(14)–C(21), 133.0(3). | ||
The pyrazole ring is planar, its main plane forms an angle of 80.9° with the C5H4 ring of the ferrocene and the N(1)–N(2) bond is located on the same direction as the Fe(η5–C5H5) moiety. As a consequence of this arrangement of groups, the N(1) atom is quite close to one hydrogen atom of the –CH2– {separation N(1)⋯H(11A) = 2.50 Å} and proximal to the H(1) and H(2) atoms of the (η5–C5H5) unit, {the distances N(1)⋯H(1) and N(1)⋯H(2) = 2.90 Å and 3.02 Å}. The two phenyl rings defined by the sets of atoms [C(15)–C(20)] and [C(21)–C(26)] are planar and their mean planes form angles of 2.6° and 63.6° with the pyrazole ring respectively.
In the crystal, the relative orientation of two neighbouring molecules with a head-to-tail disposition is such that the separation between N(1) atom of one unit and the H(20) atom of the other one (and vice versa) is 2.540 Å. This suggests the existence of two C–H(20)⋯N(1) intermolecular interactions that results in the formation of a dimer which is connected to proximal ones by C–H⋯π interactions,‡as shown in Fig. 4, b.
 | ||
| Fig. 4 a) Schematic view of the assembly of a molecule of compound 2, at (x, y, z) with a close one located at (1 − x, 1 − y, − z) through C–H⋯N interactions {atoms marked with an asterisk belong to the molecule at (1 − x, 1 − y, −z)}} and b) the connectivity of these dimeric units by different sorts of C–H⋯π intermolecular interactions. | ||
Compound 2 was also characterized in solution by mono- [1H and 13C{1H}] and two dimensional [{1H–1H} NOESY and COSY and {1H–13C}–HSQC and HMBC] NMR experiments. The most relevant features observed in the 1H–NMR spectrum was the presence of a group of two singlets of relative intensities 2:7 in the range 4.00–4.20 ppm, the less intense one is due to the pair of protons H3 and H4 of the C5H4 ring, while the other one was assigned to the remaining protons of the Fc group. The two additional singlets at δ = 6.56 and 5.09 ppm (of relative intensities 1:2) were ascribed to the proton on position 4 of the pyrazolyl ring and to those of the –CH2– unit.
Study of the reactivity of 2 in front of platinum(II)
In a first attempt to evaluate the potential coordination abilities of ligand 2 to platinum(II), its reactivity with cis-[PtCl2(dmso)2]24 was studied under different experimental conditions (Table 1 and Scheme 2).![i) cis-[PtCl2(dmso)2] {in a molar ratio 2:Pt(ii) = 1} in refluxing methanol. ii) SiO2 column chromatography. iii) cis-[PtCl2(dmso)2] and NaAcO (in a molar ratio: 2 : Pt(ii)OAc− = 1 : 1 : 2) in a mixture of toluene : methanol (5 : 1) under reflux. iv) Addition of PPh3 (in a molar ratio 5 : PPh3 = 1) in CH2Cl2 at 298 K for 1.5 h.](/image/article/2012/RA/c1ra01080h/c1ra01080h-s2.gif) | ||
| Scheme 2
i) cis-[PtCl2(dmso)2] {in a molar ratio 2:Pt(II) = 1} in refluxing methanol. ii) SiO2 column chromatography. iii) cis-[PtCl2(dmso)2] and NaAcO (in a molar ratio: 2:Pt(II)OAc− = 1:1:2) in a mixture of toluene:methanol (5:1) under reflux. iv) Addition of PPh3 (in a molar ratio 5:PPh3 = 1) in CH2Cl2 at 298 K for 1.5 h. | ||
| Entry | Reagents (molar ratios) | Solvent | T | t | Products (molar ratios) |
|---|---|---|---|---|---|
|
a A (5:1) mixture.
b The 1H–NMR spectrum of the crude material revealed the presence of 3 as the minor component.
c Since only small amounts of 3 (8 mg) were isolated from the SiO2 column, the molar ratio 4:5 is given.
d Traces of 4 (≈3 mg) were also obtained during the work up of the column.
|
|||||
| I | 2 and cis-[PtCl2(dmso)2] | MeOH | reflux | 1.0 | 3 and 4 |
| (1:1) |
(1.00:0.11) |
||||
| II | 2 and cis-[PtCl2(dmso)2] | MeOH | reflux | 3.5 | 3 and 4 |
| (1:1) |
(0.21:1.00) |
||||
| III | 2, cis-[PtCl2(dmso)2] and NaAcO | Toluene/MeOHa | reflux | 3.5 | 3 b, 4 and 5 |
| (1:1:1) |
(1.00:0.18)c |
||||
| IV | 2, cis-[PtCl2(dmso)2] and NaAcO | Toluene/MeOHa | reflux | 3.5 | 4 and 5 |
| (1:1:2) |
(0.36:1.00) |
||||
| IV | 2, cis-[PtCl2(dmso)2] and NaAcO | Toluene/MeOHa | reflux | 7.0 | 5 d |
| (1:1:2) |
|||||
| V | 5 and PPh3 | CH2Cl2 | 298 K | 1.0 | 6 |
| (1:1) |
The treatment of equimolar amounts of 2 and cis-[PtCl2(dmso)2] in refluxing methanol for 1.0 h followed by the work up of a SiO2 column chromatography afforded the trans- and cis- isomers of [Pt{1-(Fc–CH2)-3,5-Ph2–C3HN2}Cl2(dmso)] (3) and (4) respectively, in a molar ratio 3:4 =1.00:0.11 (Table 1, entry I). Longer refluxing periods (t = 3.5 h, Table 1, entry II)} also produced a mixture of the two isomers but with the molar ratio 3:4 inverted (0.21:1.00). Thus indicating that longer reaction periods induced the preferential formation of the cis- isomer (4). This finding, is consistent with previous work on the reactivity of cis-[PtCl2(dmso)2] with related N–donor ligands4a,4c,25 and has allowed us to optimize the synthesis of 4.
Compounds 3 and 4 were characterized by elemental analyses, mass spectrometry, IR and mono and two-dimensional NMR studies and product 4 was also characterized by X-ray diffraction (see below). Elemental analyses were consistent with the proposed formulae and the IR spectra of 3 and 4 showed the typical bands due to the coordinated dmso ligand.26 The assignment of the signals observed in the 1H NMR spectra of 3 and 4 was achieved with the aid of {1H–1H} NOESY and COSY experiments. 195Pt{1H} NMR spectra of 3 and 4 showed a singlet (at δ = −2998 and −2850 ppm, respectively). The variation of the chemical shift Δδ(195Pt) = δ(for 4) − δ(for 3) = 148 ppm agrees with the values reported for the trans- and cis- isomers of the related [Pt(N–donor ligand)Cl2(dmso)] complexes.4b,4c,25,27,28
The crystal structure of 4 consists of discrete molecules of [Pt{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] (Fig. 5) in which the platinum(II) is bound to the nitrogen N(1) of the pyrazolyl unit, the sulphur, S(1), of the dmso ligand and two chloride ligands {Cl(1) and Cl(2)} in a cis- arrangement {Cl(1)–Pt–Cl(2) bond angle = 90.61(5)°}. Bond lengths as well as bond angles around the platinum(II) are consistent with those reported for related cis-[Pt(N–donor ligand)Cl2(dmso)] complexes.23,25,27,29
![ORTEP plot of cis-[Pt{1-(Fc−CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] (4) showing 50% probability displacement ellipsoids. Hydrogen atoms have been omitted for clarity.](/image/article/2012/RA/c1ra01080h/c1ra01080h-f5.gif) | ||
| Fig. 5 ORTEP plot of cis-[Pt{1-(Fc−CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] (4) showing 50% probability displacement ellipsoids. Hydrogen atoms have been omitted for clarity. | ||
The three rings of the 3,5-Ph2–C3HN2 moiety are planar and the two phenyl rings, defined by the sets of atoms [C(15)–C(20)] and [C(21)–C(26)] form angles of 47.5° and 49.6°, respectively with the main plane of the heterocycle. The N(1)–N(2) bond of the pyrazole ring is directed in the opposite direction to the Fe(η5–C5H5) unit. As expected, the bond lengths and angles of the ferrocenyl unit are consistent with those of monosubstituted ferrocene derivatives,23 the two pentagonal rings are parallel (tilt angle = 0.41°) and their relative orientation deviates by ca. 8.8(8)° from the ideal eclipsed conformation.
Due to the increasing interest on platina- and palladacycles containing planar N–donor ligands due to their applications in different areas,11–17 and since it is well-known that the formation of platina- (or palladacycles) with bidentate [C(phenyl), N]− or [C(ferrocenyl), N]− ligands commonly requires the presence of a base (i.e.NaOAc or the ligand itself),4b,4c,10a,10b,25b,30 we also explored the reactivity of ligand 2 toward Pt(II) in the presence of sodium acetate.
When compound 2 was treated with equimolar amounts of cis-[PtCl2(dmso)2]24 and NaAcO in a mixture of toluene:methanol (5:1) under reflux for 3.5 h (Table 1, entry III) the formation of a platinum mirror on the walls of the Erlenmeyer flask was observed and a deep-brown solution was obtained. Concentration of the solution, followed by silica gel column chromatography, gave traces of 3, compound 4 and a new platinum(II) complex, 5. Characterization data of 5 (see the experimental section) were consistent with those expected for [Pt{3-(C6H4)-5-Ph-1-(Fc−CH2)–(C3HN2)}Cl(dmso)] (5), in which ligand 2 behaves as a monoanionic bidentate [C(phenyl), N(pyrazole)]− ligand (type B in Fig. 2) and the dmso is in a cis-arrangement to the metallated carbon atom in agreement with the transphobia effect.31
Several additional experiments were carried out in order to increase the yield of 5. Shorter refluxing periods (t) (1.0 h ≤ t < 3.5 h) produced a significant decrease of the molar ratios 5:4. and for longer reaction times (4 h ≤ t ≤ 24 h) the amount of metallic platinum formed increased, but the yield of 5 did not improve significantly.§ Better results were obtained when the reaction was performed using a two-fold excess of sodium acetate (molar ratio NaAcO:Pt(II) = 2, Table 1, entry IV and Scheme 2, step B) and long refluxing periods (t = 7.0 h), in this case 5 was the major product. Furthermore, it should be noted that the 1H–NMR spectra of the crude materials isolated in any of these reactions did not provide evidence of the presence of any other type of platinacycle suggesting that under the experimental conditions used, the activation of the σ[C(phenyl)–H] bond of 2 is preferred over that of the σ[C(ferrocenyl)–H] bond.
The results presented in this section suggest that the formation of the platinacycle 5 is a multistep process that involves the replacement of one dmso ligand of the starting material by methanol to give cis-[PtCl2(MeOH)(dmso)] which may isomerize to the trans- form. Further coordination of the N–donor ligand 2 would produce the cis- and trans- isomers of [Pt{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] respectively. Finally, the activation of the σ[C(sp2, phenyl)–H] bond of 3 would give the platinacycle 5. This in good agreement with the mechanism postulated for the cycloplatination of aryloximes and related N–donor ligands and the results obtained recently from a DFT based theoretical study of the cycloplatination process that points out that the trans- isomers of [Pt(L)Cl2(dmso)] are the key intermediates of the process.30b However, it should be noted that ligand 2 has different σ(C–H) bonds which are susceptible to metallation (Fig. 1) and although ferrocene derivatives are more prone to electrophillic attacks than their phenyl analogues,1 the formation of 5 should be ascribed to the closer orientation between the platinum(II) bound to heterocyclic nitrogen and the σ[C(phenyl)–H] rather than the σ[C(ferrocenyl)–H] bonds.
The use of molecular models for the cis- (3) and trans- (4) isomers of [Pt{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] reveals that the “PtCl2Sdmso” moiety should be nearly orthogonal to the pyrazole ring to minimize steric hindrance between the phenyl ring and the Cl or dmso ligands. This is the typical orientation found in the crystal structures of cis- or trans-[Pt(planar N–donor ligand)Cl2(dmso)].23,24,25,29 The formation of the metallacycle 5 from 3 requires the release of one of the ligands in a cis- arrangement to the σ(Pt–N) bond and the rotation of the phenyl ring (on position 3 of the pyrazole ring) to achieve a proper orientation between the σ[C(phenyl)–H] bond and the platinum(II).
Further treatment of the cycloplatinated complex 5 with the equimolar amount of PPh3 in CH2Cl2 produced [Pt{3-(C6H4)-5-Ph-1-(Fc−CH2)–(C3HN2)}Cl(PPh3)] (6) (Scheme 2, step C and Table 1, entry V) in a fairly good yield. Elemental analyses of 6 were consistent with the proposed formula. The IR spectrum suggested the binding of the PPh3 ligand to the platinum(II) and 1H, 31P{1H} and 195Pt{1H} NMR spectra indicated the presence of a [C(phenyl), N(pyrazole)]−group and a PPh3 ligand in a cis- arrangement to the metallated carbon, in agreement with the transphobia effect.31
Study of the reactivity of 2 in front of palladium(II)
In a first attempt to evaluate the potential coordination abilities of ligand 2 to palladium(II), its reactivity with cis-[PdCl2(dmso)2] was studied under different experimental conditions (Table 2 and Scheme 3).![i) Equimolar amount of [PdCl2(dmso)2], in refluxing MeOH at 298 K for 0.5 h. ii) [PdCl2(CH3CN)2] or Na2[PdCl4] {molar ratio 2:Pd(ii) = 1 or 2 (see text)} in MeOH at 298 K, 24h. iii) Equimolar amount of Pd(OAc)2 in toluene under reflux for 3.5 h followed by the addition of PPh3 in CH2Cl2 under stirring at 298 K for 1.5 h, subsequent treatment with LiCl in acetone for 2.5 h. iv) SiO2 column chromatography. v) Reaction with Na2[PdCl4] and Na(OAc)·3H2O (in a 1 : 1 : 1 molar ratio) in MeOH at 298 K for 24 h, followed by the addition of PPh3 in CH2Cl2 for 1.5 h at 298 K.](/image/article/2012/RA/c1ra01080h/c1ra01080h-s3.gif) | ||
| Scheme 3
i) Equimolar amount of [PdCl2(dmso)2], in refluxing MeOH at 298 K for 0.5 h. ii) [PdCl2(CH3CN)2] or Na2[PdCl4] {molar ratio 2:Pd(II) = 1 or 2 (see text)} in MeOH at 298 K, 24h. iii) Equimolar amount of Pd(OAc)2 in toluene under reflux for 3.5 h followed by the addition of PPh3 in CH2Cl2 under stirring at 298 K for 1.5 h, subsequent treatment with LiCl in acetone for 2.5 h. iv) SiO2 column chromatography. v) Reaction with Na2[PdCl4] and Na(OAc)·3H2O (in a 1:1:1 molar ratio) in MeOH at 298 K for 24 h, followed by the addition of PPh3 in CH2Cl2 for 1.5 h at 298 K. | ||
| Entry | Reagents (molar ratios) | Solvent | T | t | Products |
|---|---|---|---|---|---|
|
a See text.
b Yields of 8 were 35% or 89% for molar ratios 2:Pd(II) = 1 and 2, respectively.
c Followed by: i) treatment with the equimolar amount of PPh3 in CH2Cl2 at 298 K, ii) subsequent addition of LiCl in acetone and stirring of the mixture at 298 K for 2.5 h, and iii) SiO2 column chromatography.
d Small amounts of [PdCl2(PPh3)2] were also isolated as a by-product.
e Treatment with a slight excess (15%) of PPh3 in CH2Cl2 at 298 K for 1.5 h and the subsequent separation of 10 and [PdCl2(PPh3)2] by SiO2 column chromatography.
f When this reaction was performed at higher temperatures, mixtures containing 9 and 10 were isolated.
|
|||||
| I | 2 and cis-[PdCl2(dmso)2] | MeOH | reflux | 0.5 | 7 |
| (1:1) |
|||||
| II | 2 and cis-[PdCl2(CH3CN)2] | MeOH | 298 K | 24.0 | 8 |
| (1:1) or (2:1)a |
|||||
| III | 2 and Na2[PdCl4]b | MeOH | 298 K | 24.0 | 8 |
| (1:1) or (2:1) |
|||||
| IV | 2 and Pd(AcO)2c | Toluene | reflux | 3.5 | 9 d |
| (1:1) |
|||||
| V | 2, Na2[PdCl4] and NaAcO·3H2Oe | MeOH | 298 Kf | 24.0 | 10 d |
| (1:1:1) |
The reaction between equimolar amounts of 2 and cis-[PdCl2(dmso)2] in refluxing methanol for 0.5 h (Scheme 3, A and Table 2, entry I) produced a yellow solid that was identified according to its elemental analyses, mass spectra and NMR studies (see experimental section) as cis-[Pd{1-(Fc-CH2)-3,5-Ph2–(C3HN2)}2Cl2] (7). Compound 7 is similar to complex 4, except for the nature of the metal atom {M = Pt (in 4) or Pd (in 6)} and in fact X-ray studies reveal that they are isostructural.
The crystal structure of 7 consists of discrete molecules of [Pd{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] (Fig. 6). In each of these molecules the palladium(II) atom is in a slightly distorted square-planar environment and it is bound to the nitrogen N(1) of the pyrazolyl unit, two chloride ligands {Cl(1) and Cl(2)} in a cis- arrangement {Cl(1)-Pd–Cl(2) bond angle = 90.74(7)°} and the sulphur, S(1), of the dmso ligand. Bond lengths and angles around the palladium(II) (Table 3) are similar to those of related cis-[Pd(N–donor ligand)Cl2(dmso)] complexes.23,32 The two phenyl rings, defined by the sets of atoms [C(15)–C20)] and [C(21)–C(26)] are planar and form angles of 47.0° and 48.9°, respectively with the main plane of the heterocycle.
![ORTEP plot of cis-[Pd{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] (7), 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity.](/image/article/2012/RA/c1ra01080h/c1ra01080h-f6.gif) | ||
| Fig. 6 ORTEP plot of cis-[Pd{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}Cl2(dmso)] (7), 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity. | ||
| M = Pt (4) | M = Pd (7) | |
|---|---|---|
| a Average values for the Fc moiety. | ||
| Bond lengths | ||
| M–N(1) | 2.021(3) | 2.010(5) |
| M–Cl(1) | 2.2813(12) | 2.2630(17) |
| M–Cl(2) | 2.3166(10) | 2.3151(18) |
| M–S(1) | 2.2044(10) | 2.1965(19) |
| N(1)–N(2) | 1.362(3) | 1.355(9) |
| N(2)–C(11) | 1.450(4) | 1.444(11) |
| N(2)–C(12) | 1.363(4) | 1.382(10) |
| C(10)–C(11) | 1.515(5) | 1.471(13) |
| C(12)–C(19) | 1.367(5) | 1.400(8) |
| Fe–Ca | 2.039(7) | 2.04(4) |
| C–Ca | 1.41(1) | 1.41(2) |
| Bond angles | ||
| Cl(1)–M–Cl(2) | 90.61(5) | 90.74(7) |
| Cl(1)–M–S(1) | 89.94(5) | 90.04(7) |
| N(1)–M–Cl(2) | 88.84(8) | 88.35(19) |
| N(1)–M–S(1) | 90.68(8) | 90.92(19) |
| M–N(1)–N(2) | 123.29(19) | 121.8(5) |
| C(10)–C(11)–N(2) | 112.4(3) | 115.1(7) |
| C(12)–N(2)–N(1) | 108.9(2) | 106.0(6) |
| C(11)–N(2)–N(1) | 121.1(2) | 124.8(7) |
| N(2)–C(12)–C(13) | 122.4(3) | 124.7(7) |
As expected, the bond lengths and angles of the ferrocenyl unit are consistent with those of monosubstituted ferrocene derivatives.23 The two pentagonal rings are parallel (tilt angle = 0.41°) and their relative orientation deviates by 8.8(8)° from the ideal eclipsed one. In this compound the orientation of the pyrazolyl ring and the Fe(η5–C5H5) unit it is similar to that of 4, suggesting that the formation of 4 or 7 requires rotation around the N–CH2– or the Fc–CH2– bonds to reduce steric effects.
When the reaction was carried out using equimolar amounts of ligand 2 and cis-[PdCl2(CH3CN)2] in methanol for 24 h (Table 2, entry II and Scheme 3, step B), a yellow solid was obtained and its characterization data (see experimental section) was consistent with those expected for the trimetallic complex trans-[Pd{1-(Fc–CH2)-3,5-Ph2–(C3HN2)}2Cl2] (8). The yield of this synthesis was low but it improved considerably when a two fold excess of 2 was used. Compound 8 was also isolated by treatment of the ligand and Na2[PdCl4] {in molar ratios (1:1) or (2:1)} in methanol at room temperature (Table 2, entry III).
It should be noted that no evidence of the presence of any other type of palladium(II) complex was detected by 1H–NMR studies of the crude (as well as of the mother liquors) of any of the experiments presented in Table 2, entries I–III.
In view of these results and in a further attempt to achieve cyclopalladated complexes, we decided to use more severe experimental conditions based on the reaction of the ligand 2 and Pd(AcO)2 (in a 1:1 molar ratio) in refluxing toluene for 3.5 h (Scheme 3, C), subsequent treatment of the crude with a slight excess of PPh3 (in CH2Cl2) followed by LiCl (in acetone) (Scheme 3, step C). Evaporation of the solvent produced a brownish residue, which was later on purified by silica gel column chromatography. The use of CH2Cl2 as eluant released a band that was collected and concentrated to dryness giving a bright yellow solid 9, whose elemental analyses (see the experimental section) were consistent with those expected for a palladacycle of the type: [Pd(C,N)Cl(PPh3)]. Mono- and two dimensional NMR spectra revealed that 9 contains: a) a five-membered palladacycle in which compound 2 acts as a [C(phenyl), N(pyrazolyl)]− ligand (Fig. 2, type A), and b) the PPh3 ligand occupies the adjacent position to the metallated carbon atom in the coordination sphere of the palladium(II). All these findings suggested that 9 was [Pd{3–(C6H4)–5-Ph-1-(Fc-CH2)–(C3HN2)}Cl(PPh3)] (9), which is similar to complex 6, except for the nature of the metal atom.
X-ray diffraction studies revealed that the crystals contained a 1:1 array of molecules of CH2Cl2 and [Pd{3–(C6H4)-5-Ph-1-(Fc–CH2)–(C3HN2)}Cl(PPh3)] (9). In each one of these heterodimetallic units (Fig. 7 and Table 4), the palladium(II) is in a slightly distorted square-planar environment, where it is bound to the heterocyclic nitrogen of the pyrazole ring and the C(26) atom, thus confirming that metallation occurred on the phenyl ring on position 3 of the heterocycle. The two remaining coordination sites are occupied by a chloride and the phosphine ligand. The value of the P–Pd–C(26) bond angle {93.18(8)°} indicates that the PPh3 ligand is in a cis position in relation to the Pd–C bond, in agreement with the transphobia effect.31 Bond lengths and angles around the palladium(II) (Table 4) are similar to those reported for related palladacycles of the type [Pd{C(sp2, phenyl),N}Cl(PPh3)].23
![ORTEP plot of compound [Pd{[3-(C6H4)–5-Ph-1-{(η5–C5H5)Fe(η5–C5H4)–CH2}–(C3HN2)]}Cl(PPh3)] (9), 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity.](/image/article/2012/RA/c1ra01080h/c1ra01080h-f7.gif) | ||
| Fig. 7 ORTEP plot of compound [Pd{[3-(C6H4)–5-Ph-1-{(η5–C5H5)Fe(η5–C5H4)–CH2}–(C3HN2)]}Cl(PPh3)] (9), 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity. | ||
| 9 | 10 | |
|---|---|---|
| a This refers to the C(26) atom of complex 9 or to the C(1) atom of 10. b Average value for the Fc unit. | ||
| Bond lengths | ||
| Pd–P | 2.2492(12) | 2.2314(14) |
| Pd–Cl | 2.4007(14) | 2.3907(14) |
| Pd–N(1) | 2.122(2) | 2.128(3) |
| Pd–Ca | 2.043(3) | 2.017(4) |
| N(1)–N(2) | 1.365(2) | 1.339(4) |
| N(2)–C(11) | 1.477(3) | 1.371(5) |
| C(11)–C(10) | 1.500(4) | 1.521(6) |
| Fe–Cb | 2.037(7) | 2.04(2) |
| C–Cb | 1.41(2) | 1.43(1) |
| Bond angles | ||
| N(1)–Pd–Ca | 80.46(10) | 91.53(15) |
| N(1)–Pd–Cl | 97.13(7) | 84.63(10) |
| P–Pd–Cl | 91.05(5) | 94.20(5) |
| P–Pd–Ca | 93.18(8) | 91.08(12) |
In the crystal, two molecules of [Pd{3–(C6H4)–5-Ph-1-(Fc–CH2)–(C3HN2)}Cl(PPh3)] are assembled in pairs by C–H⋯π interactions involving the hydrogen atom H(29) of a molecule and the ring defined by the set of atoms [C(39)–C(44)] [the distance H(29)⋯centroid of this ring is 3.491 Å] of the other one and vice versa. In addition, in each one of these units, the separation between the Cl(1) atom and the H(45A) atom of the CH2Cl2 molecule (2.837 Å) suggests a weak C–H⋯Cl interaction.
For ligand 2, the activation of any of the two ortho σ[C(ferrocene)–H] bonds of the Fc unit would give the six-membered metallacycles with a σ[Pd–C(ferrocene)] bond (B-type in Fig. 1) but none of the reactions studied here allowed us to isolate this sort of product. In view of this, and since it is well-known that one of the most common methods used to obtain palladacycles with [C(ferrocene),N]− ligands consists of the treatment of the ferrocenyl ligand, M2[PdCl4] (M = Li+ or Na+) and NaAcO·3H2O, in methanol at room temperature,17b,33,34 we also decided to explore whether for ligand 2 this procedure could allow us to isolate metallacycles with a σ[Pd–C(ferrocene)] bond.
Reaction of 2 with Na2[PdCl4] and NaAcO·3H2O in methanol at room temperature for 24 h produced a brown solid (Scheme 3, D). Further treatment with PPh3 in dichloromethane and the subsequent purification of the crude by column chromatography on silica gel gave an orange solid 10. Its elemental analyses were consistent with those expected for a palladacycle of the type [Pd(C,N)Cl(PPh3)], but its 1H and 31P{1H}–NMR spectra were markedly different from those of 9. The position of the singlet detected in the 31P{1H} NMR spectrum (δ = 38 ppm) was similar to those reported for related compounds with N–donor ferrocenyl ligands and a σ[Pd–C(sp2,ferrocene)] bond (35 ppm < δ < 40 ppm),33 and was up-field shifted when compared with 9 (δ = 46 ppm). Its 1H–NMR spectrum was more complex than expected and showed six singlets with relative intensities (1:1:2:1:1:1) in the range 3.0 < δ < 4.2 ppm. This pattern, which is markedly different from those of compounds 3–9 or of five-membered palladacycles formed by metallation of the (η5–C5H4) ring of the Fc group,33 suggests the presence of a σ(Pd–C) bond generated by a transannulation process.
The X-ray crystal structure of 10 (Fig. 8) revealed that the palladium(II) was bound to the nitrogen atom N(1) and the C(1) atom of the unsubstituted (η5–C5H5) ring of 2. Two remaining coordination sites are occupied by a chloride, and the phosphorous atoms of the PPh3 ligand, which is in the adjacent position to the Pd–C bond in agreement with the transphobia effect.31
![ORTEP plot of compound [Pd{1-[(η5–C5H4)Fe(η5–C5H4)–CH2]-3,5-Ph2-1-(C3HN2)}Cl(PPh3)] (10, 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity.](/image/article/2012/RA/c1ra01080h/c1ra01080h-f8.gif) | ||
| Fig. 8 ORTEP plot of compound [Pd{1-[(η5–C5H4)Fe(η5–C5H4)–CH2]-3,5-Ph2-1-(C3HN2)}Cl(PPh3)] (10, 50% probability displacement ellipsoids are shown and hydrogen atoms have been omitted for clarity. | ||
As a consequence of the different mode of binding, the separation between the two metal centres (3.563 Å) is smaller than in 9 (5.358 Å) or in the coordination complexes 5 and 7, but it clearly exceeds the sum of the van der Waals radii of these two atoms.35 This precludes the existence of any direct interaction between iron and palladium.
The average values of the Fe–C and C–C bond lengths of the Fc moiety are similar to those reported for related 1,1′ disubstituted ferrocene derivatives.23 Moreover, the two pentagonal rings of this unit are planar, parallel (tilt angle of 1.44°) and they deviate by ca. 9° from the ideal eclipsed conformation. The pyrazole ring is planar and it is nearly orthogonal to the rings of the Fc unit (angles between their mean planes = 89.0° and 90.0°). This arrangement of substituents is very similar to that of the free ligand¶ and differs from those found in 7 and 9 in which the pyrazole and the Fe(η5–C5H5) unit are on opposite sides of the coordination plane of the metal.
According to the mechanism accepted for the cyclopalladation and cycloplatination of N– donor ligands, the reaction proceeds through coordination of the metal(II) ion to the nitrogen followed by the subsequent electrophillic attack at the carbon atom.36 Once the M–N bond is formed the relative orientation between the metal centre and the σ(C–H) bonds susceptible to metallation appears to be particularly relevant in determining the regioselectivity of the process.
It should be noted that since: a) previous theoretical studies revealed that six-membered palladacycles are expected to be more stable than those containing five-membered ring37 and b) compound 10 was isolated under milder experimental conditions (room temperature) than 9 (under reflux and using longer reaction periods), we tentatively propose that 10 arises from kinetic control while 9 is formed under thermodynamic control. In order to confirm this hypothesis, we decided to investigate the reaction between equimolar amounts of ligand 2, Na2[PdCl4] and NaAcO·3H2O in methanol for 24 h, followed by the treatment of the raw material with PPh3. When the reaction was carried out at room temperature the 1H–NMR spectrum of the crude revealed the presence of 10 and 9 in a molar proportion 6.0:0.8 and when the reaction was performed under reflux during the same period of time the molar ratio 10:9 was 2.3:1.0. This suggests that the proclivity of 2 to undergo the transannulation process to give 10 decreases upon heating when compared with metallation of the phenyl ring to produce 9.
It should be noted that despite this, it is well-known that: a) ferrocene derivatives are more prone to electrophillic attacks than their benzene analogues1 and b) pallada- and platinacycles derived from the metallation of the substituted (η5–C5H4) ring of the Fc moiety are known,4,33,34 and in the case of ligand 2 the presence of this type of six-membered palladacycles were not detected in any of the reactions investigated.
Theoretical studies
In a first step to rationalize the results obtained in the reactions of 2 with palladium(II) salts or complexes, we decided to undertake DFT calculations38 for the three different types of metallacycles, A–C, depicted in Fig. 2 that arise from the metallation of the phenyl (A), the C5H4 ring (B) or formed by a transannulation process (C). The models used contained a chloride and an additional neutral L ligand (in a cis- arrangement to the metallated carbon) and also two different L ligands (PH3 and dmso) in order to find out if the nature of this group could induce significant variations in the stability of the complexes. The choice of PH3 instead of the PPh3 ligand present in the palladium(II) complexes 9 and 10 was done in order to simplify the calculations and to minimize computational time.For these six model compounds, the geometries were optimized before the calculations and their total energy was determined. The results (Table 5) obtained showed that the total energy of type A molecules was smaller than those of their isomers arising from the metallation of any of the two rings of the Fc unit (types B or C). For compounds with L = PH3 the total energy of the molecule increased according to the sequence A < C < B and an identical trend was found for their analogues with dmso. Consequently, from a theoretical point of view, the formation of A-type palladacycles with a σ[Pd–C(sp2, phenyl)] bond appears to be strongly favoured in the gas phase.
Since the polarity of the solvents used was different in the preparation of 9 and 10, we carried out additional computational studies in order to elucidate whether the solvent (toluene or methanol) could affect the relative stability of the three models. As can be easily seen in Table 5, in both solvents the minimum energy was obtained for A-type palladacycles and, for an identical neutral ligand, the energy of model B was again higher than that of its C-type analogue. When toluene is replaced by methanol the relative total energy of models B and C decreased, thus indicating that the polarity of the solvent may play a key role.
In a further step and in order to modelize the isolated products, we also calculated the total energies of complexes containing PPh3. This family of products shows the same stability trend (Table 5) as the simpler models with L = PH3 or dmso, and again the replacement of toluene by methanol reduces the differences ΔEr = [ET (for B or C types) − ET (for A-model)]. Thus, according to these computational studies the cyclometallation of the phenyl ring is strongly preferred over the transannulation process or the metallation of the (η5–C5H4) unit.
In view of this, we focused our attention in other factors that could affect these metallation processes. First, structural studies have shown that the orientation of the pyrazole ring in the free ligand 2 and in complex 10 is similar.¶ This is in agreement with the results obtained for 4-ferrocenyl-1,3-oxazolidine (11) and its palladium(II) complex formed by metallation of the (η5–C5H5) ring.4a Second, in 9 (as well as in the coordination compounds 4 and 7, which were also isolated under refluxing conditions) the heterocycle is in the opposite direction to Fe(η5–C5H5). This requires a change of the conformation of the ligand before or after the formation of the N–metal bond (Pd or Pt). Consequently, we decided perform molecular mechanics calculations to evaluate the variation of the total energy of ligand 2 for different orientations of the (η5–C5H4) ring of the Fc moiety and the heterocycle using the MM3 method.39 Such arrangements of substituents were generated by modifying the torsion angles defined by the atoms C2Fc–C1Fc–C–N2 and C1Fc–C–N2–N1 {hereinafter referred to as Φ(1) and Φ(2), respectively} in the range: [−180°, +180°].
The conformational map shows that if the free ligand is to achieve the same orientation as in 9 (or products 4 or 7), prior to the binding of the metal, this requires the change of Φ(1) and Φ(2) and an energy input. This would be more likely to occur if the reactions are performed under reflux (as happens in the preparation of 4, 7 and 9).
On the other hand, 9 and 10 were isolated using different starting reagents {Na2[PdCl4] or Pd(AcO)2}, and then the first step of the process consists of the coordination of the PdCl![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) moiety (for 10) or of Pd(AcO) (for 9) to the nitrogen of ligand 2. In the former case, where the reaction is performed in methanol at 298 K: a) according to the calculations, the difference between the relative total energy of the A and C–types decreases and b) the change of the conformation of the ligand requires an energy input and appears to be less likely to occur; consequently, once the Pd–N bond is formed the σ(C–H) bond of the C5H5 ring is extremely close to the metal centre and has the proper orientation as to undergo activation, this would lead to the C–type metallacycles.
moiety (for 10) or of Pd(AcO) (for 9) to the nitrogen of ligand 2. In the former case, where the reaction is performed in methanol at 298 K: a) according to the calculations, the difference between the relative total energy of the A and C–types decreases and b) the change of the conformation of the ligand requires an energy input and appears to be less likely to occur; consequently, once the Pd–N bond is formed the σ(C–H) bond of the C5H5 ring is extremely close to the metal centre and has the proper orientation as to undergo activation, this would lead to the C–type metallacycles.
In contrast with these findings, the first step of the formation of 9 consists of the binding of the Pd(AcO) unit, (with a greater effective bulk than the PdCl unit) and in order to minimize steric effects, the conformation of 2 should change. This may be promoted at high temperatures (under reflux). A similar argument can be used to explain the variation observed in the arrangement of the ferrocene and pyrazole units in cis-[M[1-{(η5–C5H5)Fe(η5–C5H4)–CH2}-3,5-Ph2–C3HN2]Cl2(dmso)] {M = Pt (4) or Pd (7)} when compared with those of 2 or 9.
For this arrangement of groups the σ[C(phenyl)–H] bond on the ortho site is closer to the M(II) centre and has a better orientation than any of the σ[C(sp2, ferrocene)–H] bonds of the (η5–C5H5). Thus the cyclometallation of the phenyl ring is strongly preferred over the transannulation process.
Conclusions
The new 3,5-diphenylpyrazole derivative [1-(Fc−CH2)-3,5-Ph2–(C3HN2)] {Fc = (η5–C5H5)Fe(η5–C5H4)−} (2) with a Fc–CH2– unit on position 1 of the heterocycle has been prepared and characterized. The study of its reactivity in front of platinum(II) and palladium(II) salts has lead to heterodi- (3–7, 9 and 10) or heterotrimetallic complexes (8) complexes, where 2 acts as a neutral N– donor ligand (in 3, 4, 7 and 8) or as a bidentate [C(sp2,phenyl),N]− (in 5 and 6 or 9) or [C(sp2, ferrocene),N]− in 10.Furthermore, we have demonstrated that the proper selection of the initial Pd(II) starting material {cis-[PdCl2L2] (L =dmso or CH3CN), Na2[PdCl4] or Pd(AcO)2} and the experimental conditions {temperature, solvents (methanol or mixtures of toluene/methanol); the presence or absence of additional NaAcO} allows the control of: a) the type of final product {heterodi- (in 7, 9 and 10) or trimetallic (in 8)}, b) the mode of binding of the ligand {monodentate (in 7 and 8) or bidentate (C,N)− (in 9 and 10)} and c) the regioselectivity of the cyclometallation process and the type of σ(Pd–C) bond formed {σ[Pd–C(sp2, phenyl)] (in 9) or σ[Pd–C(sp2,ferrocene)] (in 10)}.
The formation of compound 10 bond takes place under milder experimental conditions than that of the metallacycle 9 with a five-membered ring and a σ[Pd–C(sp2,phenyl)] bond. The computational studies undertaken for the simplified models of the palladacycles (A–C) indicate that complexes formed by metallation of the phenyl ring (A-type) are more stable than type C (transannulation products) and B models (with a 1,2-disubstituted ferrocene unit) that are the less stable products.
In addition, the studies summarized here provide conclusive evidences of the crucial role of the solvent, the temperature and the steric effects induced by the palladium(II) or platinum(II) salt or complex used as starting material in determining the nature of the C–H bond to be activated. Both theoretical and experimental data suggest that: a) the formation of the metallacycles with a σ{M–C(sp2, phenyl)} bond (5, 6, and 9 with A-types cores) is strongly favoured at high temperatures (refluxing toluene or methanol or mixtures of both) which may provide the energy input required to change the orientation of the ligand and when the starting reagents are bulky {[MCl2(dmso)2] or Pd(AcO)2}; b) the activation of the C–H bond of the unsubstituted (η5–C5H5) ring, to form 10 (C–type) is favoured at low temperature (methanol) and c) metallacycles with a 1,2-(η5–C5H3) ring (B-type cores) are the less stable ones in both solvents and have not been obtained in any experiment.
Finally, since it is well-known that palladium(II) and platinum(II) complexes containing bidentate (C,N)− ligands have a variety of applications in several fields (i.e. organometallic synthesis and homogeneous catalysis) and are attractive in view of their potential photo-optical properties or biological activities, the new products presented here appear to be excellent candidates to be studied in these fields.
Experimental
Materials and methods
FcCH2OH and the complexes [MCl2(dmso)2] (M = Pd or Pt) were prepared as reported previously.20,24,40 The remaining reagents were obtained from commercial sources and used as received. Except methanol (which was HPLC-grade), the remaining solvents were dried and distilled before use.41 Elemental analyses were carried out at the Serveis de Cientifico-Tècnics (Universitat Barcelona). Mass spectra (ESI+) were performed at the Servei d'Espectrometria de Masses (Universitat de Barcelona). Infrared spectra were obtained with a Nicolet 400FTIR instrument using KBr pellets. The Rf values were obtained using SiO2 (Merck silica gel 60 F254) plates and the solvents specified in the characterization section of the corresponding product. High resolution 1H–NMR spectra and the two-dimensional {1H–1H}–NOESY and COSY experiments were registered with a Varian VRX-500 or a Bruker Avance DMX-500 MHz instruments. 195Pt{1H}–NMR spectra of compounds 3–6 were obtained with a Bruker-250 MHz instrument. 31P{1H}–NMR spectra of 6, 9 and 10 were recorded with a Varian-Unity-300 instrument. In all cases the solvent for the NMR experiments was CDCl3 (99.9%) and the references were SiMe4 [for 1H NMR], P(OMe)3 [δ(31P) = 140.17 ppm] for 31P–NMR and H2[PtCl6] [δ195Pt{H2[PtCl6]} = 0.0 ppm] for 195Pt{1H}–NMR experiments. All these NMR studies were performed at 298 K. The chemical shifts (δ) are given in ppm and the coupling constants (J) in Hz. In the characterization section of each product, the assignment of the signals detected in the 1H–NMR spectra refer to the labelling patterns presented in Schemes 1, 2 and 4. UV-vis. spectra of 1.0 × 10−4 M solutions of the compounds in CH2Cl2 were recorded with a Cary 100 scan Varian UV spectrometer at 298 K.[1-{(η5–C5H5)Fe(η5–C5H4)–CH2}-3,5-Ph2–C3HN2] (2). 3,5-Diphenylpyrazole (0.5 g, 2.27 × 10−3 mol) and Fc–CH2OH (0.49 g, 2.27 × 10−3 mol) were dissolved in 50 mL of CH2Cl2. Then a solution formed by 9 mL of HBF4 and 6 mL of H2O was added dropwise under stirring. The resulting mixture was magnetically stirred at 298 K for 1 h and afterwards it was treated with 100 mL of H2O. The mixture was extracted with two 50 mL portions of CH2Cl2 and the organic layer was then dried over Na2SO4 and concentrated. Further purification by flash chromatography on silica gel using CH2Cl2 as eluant produced the release of an orange band that was concentrated to dryness on a rotary evaporator. The solid formed was collected and dried in vacuum for 24 h. (Yield: 0.91 g, 96%). 1H–NMR data: δ = 4.06 (s, 2H, H2Fc and H5Fc), 4.12 (s, 7H, H3Fc, H4Fc and Cp), 5.09 (s, 2H, –CH2–), 6.56 (s, 1H, H4), 7.33 (br, 2H, H4a and H4b), 7.40–7.70 (br.m, 6H, H3a, H5a, H2b, H3b, H5b and H6b) and 7.95 (dd, 2H, J = 8 and 1, H2a and H6a). IR (solid, cm−1): 1969(w), 1774(w), 1723(w), 1637(w), 1602(m), 1547(m), 1514(s), 1484(s), 1460(s), 1368(s), 1313(s), 1236(m), 1193(m), 1103(s), 1079(s), 997(s), 957(m), 820(s), 766(s), 703(s), 565(m) and 495(s). UV-vis. data: λ in nm (ε in M−1 cm2) = 244 (42250), 246 (42140), and 438 (4850). Rf (in CH2Cl2) = 0.88. MS (ESI+): m/z = 419.1 {[M] + H}+. Elemental analysis calc. for C26H22N2Fe; C 74.65, H 5.30, N, 6.70 (6.69), found: C 74.53, H 5.27, N 6.69.
Trans-[Pt[1-{(η5–C5H5)Fe(η5–C5H4)–CH2}-3,5-Ph2–C3HN2]Cl2(dmso)] (3). Cis-[PtCl2(dmso)2] (249 mg, 5.90 × 10−4 mol) was suspended in 30 mL of methanol and refluxed until complete dissolution. Then the hot solution was filtered out and the filtrate was poured on a solution containing 247 mg (5.90 × 10−4 mol) of 2 and 5 mL of methanol. The resulting reaction mixture was refluxed for 1 h. After this period, the solution was filtered out, the resulting filtrate was concentrated to dryness on a rotary evaporator and the brownish residue was air dried for 24 h. After this period the solid was dissolved in the minimum amount of CH2Cl2 (≈15 mL) and passed through a SiO2 column (8.5 cm × 3.0 cm). Elution with CH2Cl2 released two yellow bands that were collected and concentrated to dryness on a rotary evaporator. The first eluted band gave after concentration to dryness on a rotary evaporator the cis- isomer (4) (≈39 mg). Identical work up of the second eluted band, produced compound 3 (351 mg, 78%) as an orange-yellow microcrystalline material. 1H–NMR data: δ 3.68 [s, 6H, 3JPt,H = 19, Me(dmso)], 4.03 (s, 2H, H3Fc and H4Fc), 4.14 (s, 5H, Cp), 4.30 (s, 2H, H2Fc and H5Fc), 5.89 (s, 2H, –CH2–), 6.45 (s, 1H, H4), 7.32–7.60 (m, 8H, H3a–H5a and H2b–H6b) and 8.76 (d, 2H, J = 7.8, H2′′ and H6′′). 195Pt{1H}–NMR: δ −2998. IR (solid, cm−1) 1638(s), 1553(w), 1481(m), 1459(m), 1383(m), 1297(w), 1149(s), 1105(m), 1022(s), 920(w), 817(w), 761(m), 697(s) and 482(s) cm−1. UV-vis. data: λ in nm (ε in M−1 cm2) = 242 (36870). Rf (in CH2Cl2) = 0.64. MS (ESI+): m/z = 780.1{[M] + (NH4)}+. Elemental analysis calc. for C28H28N2OSCl2FePt C 44.11, H, 3.70, N, 3.67, S, 4.21, found, C 44.13, H 3.71, N 3.67 3.59, S 4.18.
Cis-[Pt[1-{(η5–C5H5)Fe(η5–C5H4)–CH2}-3,5-Ph2–C3HN2]Cl2(dmso)] (4). This compound was also obtained as a by-product during the preparation of 3 (see preceding section) but the method described here allows its isolation in a higher yield. The procedure is identical to that described above for the trans- isomer (3), except that the refluxing period was 3.5 h. The first eluted band produced after concentration 272 mg of compound 4 and small amounts (58 mg) of 3 were also isolated from the band collected afterwards. Crystals suitable for X-ray analyses were obtained by slow evaporation of a saturated CH2Cl2 solution of 4 at 270 K. 1H–NMR data: δ 2.43 [s, 3H, 3JPt,H = 20, Me(dmso)], 2.79 [s, 3H, 3JPt,H = 20, Me(dmso)], 3.67(s, 5H, Cp), 4.06 (s, 1H, H4Fc), 4.10 (s, 1H, H3Fc), 4.15 (s, 1H, H2Fc), 4.20 (s, 1H, H5Fc), 5.51 (d, 1H, 2J = 15.0, –CH2–), 6.06 (d, 1H, 2J = 15.0,–CH2–), 6.49 (s, 1H, H4), 7.45–7.78 (br. m, 8H, H3a–H5a and H2b–H6b) and 8.21(d, 2H, 3J = 7.8, H2a and H6a). 195Pt{1H}–NMR data: δ = −2850. IR (solid, cm−1) data: 1637(s), 1553(w), 1481(m), 1449(m), 1385(m), 1311(w), 1149(s), 1105(mn), 1075(m), 921(w), 821(w), 760(m), 699(s) and 482(s). UV-vis. data: λ in nm (ε in M−1 cm2) = 237 br.(39920). Rf (in CH2Cl2) = 0.81. MS (ESI+): m/z = 780 {[M] + (NH4)}+ and 726 {[M]–Cl}+. Elemental analysis calc. for C28H28N2OSCl2FePt: C 44.11, H 3.70, N, 3.62, S 4.21, found: C 44.03, H 3.67, N 3.57; S 4.01.
[Pt{[3–(C6H4)-5-Ph-1-{(η5–C5H5)Fe(η5–C5H4)–CH2}–C3HN2]Cl(dmso)] (5). Ligand 2 (247 mg, 5.90 × 10−4 mol) and cis-[PtCl2(dmso)2] (249 mg, 5.90 × 10−4 mol), were suspended in 25 mL of toluene. Then a solution formed by 100 mg (1.18 × 10−3 mol) of NaAcO and 5 mL of methanol was added and the mixture was refluxed for 3.5 h. After this period the hot solution was carefully filtered out, the filtrate was concentrated to dryness on a rotary evaporator and the residue was air-dried for 24 h. Afterwards, the raw material was dissolved in the minimum amount of CH2Cl2 (≈15 mL) and passed through a SiO2 column (8.5 cm × 3.0 cm). Elution with CH2Cl2 released two yellow bands that were collected and concentrated to dryness on a rotary evaporator. The first eluted band gave, after concentration to dryness on a rotary evaporator, traces (≈3 mg) of the cis- isomer (4). Identical work up of the second eluted band, produced compound 5 (269 mg) as a bright yellow-microcrystalline material. 1H–NMR data: δ = 2.79 [s, 3H, 3JPt,H = 20, Me(dmso)], 2.87 [s, 3H, 3JPt,H = 19, Me(dmso)], 3.90 (s, 5H, Cp), 4.01 (s, 1H, H4Fc), 4.07 (s, 1H, H3Fc), 4.19 (s, 1H, H5Fc), 4.90 (s, 1H, H2Fc), 5.20 (d, 1H, 2J = 15.2, –CH2–), 5.81 (d, 1H, 2J = 15.2, –CH2–), 6.40 (s, 1H, H4), 6.60 (d, 1H, J = 7.5 and 3JPt,H = 39.4, H3a), 6.95 (m, 1H, H4a), 7.45–7.78 (br. m, 6H, H5a and H2b–H6b) and 8.05 (d, 2H, H6a). 195Pt{1H}–NMR data: δ −3875. IR (solid, cm−1) 1683 (m), 1532(m), 1482(m), 1449(m), 1383(m) 1291(m), 1149(s), 1104(m), 1023(s), 999(m), 822(m), 763(2), 699(m), 496 (w), 481(m) and 436(m). UV-vis. data: λ in nm (ε in M−1 cm2) = 243 (49321), 272 (45920) and 355 (5931). Rf (in CH2Cl2) = 0.68). MS (ESI+): m/z = 698{[M]–Cl}+. Elemental analysis calc. for C28H27ClN2FeOPS: C 46.32, H 3.75, N 3.86, S 4.42, found: C 46.25, H 3.8, N 3.7, S 4.35.
[Pt{[3-(C6H4)-5-Ph-1-{(η5–C5H5)Fe(η5–C5H4)–CH2}–C3HN2]Cl(PPh3)] (6). Compound 5 (88 mg, 1.2 × 10−4 mol) was dissolved in 50 mL of CH2Cl2 and then an equimolar amount (32 mg) of PPh3 was added. The resulting mixture was stirred at 298 K for 1 h and then the undissolved materials were removed by filtration and discarded. The filtrate was then concentrated to ca. 5 mL and passed through a short SiO2–column (1.0 cm × 2.5 cm) to remove the free dmso. Elution with a CH2Cl2
:MeOH (100:0.1) mixture produced the release of a bright yellow band that was collected and concentrated to dryness on a rotary evaporator. The solid formed was collected, air-dried and then dried in vacuum for 2 days. (Yield: 95 mg, 87%). 1H–NMR data: δ 3.85 (s, 2H, H2Fc, H5Fc) 4.00 (s, 2H, H3Fc, H5Fc), 4.05 (s, 5H, Cp), 6.12 (s, 2H, –CH2–), 6.35 (td, 1H, J = 7.5 and 1.5 H5a), 6.40 (br, 2H, H4 and H3a), 6.85 (td, 1H, J = 7.5 and 1.5, H4”), 7.32–8.15 (br m, 21H, H6a, H2b–H6b and aromatic protons of the PPh3 ligand). 31P{1H}–NMR data: δ = 16.7 (d, 1JP,Pt = 4180). 195Pt{1H}–NMR data: δ −4226 (1JPt,P = 4180). IR (solid, cm−1): 1615(w), 1585(w), 1543(w), 1480(s), 1433(s), 1420(w), 1390(w), 1189(w), 1095(s), 1033(m), 810(w), 784(s), 693(s) and 520(broad) cm−1. UV-vis. data: λ in nm (ε in M−1 cm2) = 239 (48761), 275(sh) and 342(6538). Rf [in CH2Cl2: MeOH (100:0.2)] = 0.86. MS (ESI+): m/z = 873.6 {[M]–Cl}+. Elemental analysis calc. for C44H36Cl2N2FePPt: C 58.07, H 3.99, N, 3.08; found C 57.95, H 3.99 4.05; N, 2.95.
Cis-[Pd[1-{(η5–C5H5)Fe(η5–C5H4)–CH2}-3,5-Ph2–(C3HN2)]Cl2(dmso)] (7). A solution formed by cis-[PdCl2(dmso)2] (197 mg, 5.90 × 10−4 mol), ligand 2 (247 mg, 5.90 × 10−4 mol) and methanol (30 mL) was refluxed for 30 min. After this period, the solution was filtered out, the resulting filtrate was concentrated on a rotary evaporator to 10 mL. The solid formed was collected by filtration and air-dried for 24 h Afterwards it was dissolved in the minimum amount of CH2Cl2 and the resulting bright yellow solution was kept at 270 K for one day. The yellow crystals formed were collected by filtration and dried in vacuum for two days. (Yield: 321 mg, 81%). 1H–NMR data: δ 2.25 [s, 3H, Me(dmso)], 2.78 [s, 3H, Me(dmso)], 3.82 (s, 5H, Cp), 4.08 (s, 1H, H3Fc), 4.10 (s, 1H, H4Fc), 4.16 (s, 2H, H2Fc), 4.95 (s, 1H, H5Fc), 5.48 (d, 1H, 2J = 15.0, –CH2–), 5.92 (d, 1H, 2J = 15.0, –CH2–), 6.10 (s, 1H, H4), 8.32 (d, J = 7.5, 2H, H2a and H6a),7.30–7.90 (8H, H3a–H5a and H2b–H5b). IR (solid, cm−1) 1587(w), 1474(m), 1446(m), 1385(m), 1326(m), 1156(w), 1104(m), 1075(m), 819(w), 778 (m), 757(s), 700(s) and 484(m) cm−1. UV-vis. data: λ in nm (ε in M−1 cm2) = 239br. (43921). MS (ESI+): m/z = 636{[M]–Cl}+. Elemental analysis calc. for C28H28Cl2N2FeOPdS C, 49.91, H 4.19, N 4.16, S 4.76; found: C 50.0, H 4.2, N 4.05, S, 4.6).
Trans-[Pd{1-[(η5–C5H5)Fe(η5–C5H4)–CH2]-3,5-Ph2–(C3HN2)}2Cl2] (8). This compound was obtained using two alternative procedures that differ in the nature of the starting palladium(II) complex used as reagent: cis-[PdCl2(CH3CN)2] or Na2[PdCl4] {methods a) and b), respectively}.
Method a). A 300 mg amount (7.18 × 10−4 mol) of ligand 2 was dissolved in the minimum amount of MeOH. Then a solution formed by 101 mg (3.59 × 10−4 mol) of [PdCl2(CH3CN)2] and 30 mL of methanol was added. The resulting mixture was stirred at 298 K for 24 h and after this period the yellow solid formed was collected by filtration, air-dried and finally dried in vacuum for one day. (Yield: 305 mg, 84%).
Method b). A solution containing Na2[PdCl4] (105 mg , 3.57 × 10−4 mol) and 10 mL of methanol was treated with a mixture formed by ligand 2 (300 mg, 7.18 × 10−4 mol) and 5 mL of methanol. The reaction mixture was kept under continuous stirring for 24 h and the solid formed was then collected by filtration and dried as in Method a). (Yield: 324 mg, 89%).1H–NMR data: δ = 4.07 [br.s, 4H, 2(H2Fc and H5Fc)], 4.11 (s, 10H, 2 Cp), 4.22 [br.s, 4H, 2(H3Fc and H4Fc)], 5.92 (s, 4H, 2 –CH2–), 6.35 (s, 2H, 2 H4), 7.30–7.89 [br. m, 2(H2b–H6b and H3a–H5a)] and 8.09 [br, 4H, 2(H2a and H6a)]. IR (solid, cm−1) 1637(s), 1557(w), 1483(s), 1447(s), 1373(m), 1325(m), 1174(w), 1103(mn), 1040(m), 1028(m), 810(w), 765(s), 751(s), 700(s) 507(w) and 496(s) cm−1. UV-vis. data: λ in nm (ε in M−1 cm2) = 239br. (62391). MS (ESI+): m/z = 976{[M]–Cl}+ and 470.5 {[M]–2Cl}2+/2. Elemental analysis calc. for C52H44Cl2N4Fe2Pd: C 61.60, H 4.37, N 5.53; found, C 61.55, H 4.30, N 5.60.
[Pd{[3-(C6H4)-5-Ph-1-{(η5–C5H5)Fe(η5–C5H4)–CH2}–(C3HN2)]Cl(PPh3)] (9). Pd(OAc)2 (161 mg, 7.23 × 10−4 mol) was added to a solution containing 300 mg (7.18 × 10−4 mol) of 2 and 20 mL of toluene and the mixture was refluxed for 3.5 h. After this period the resulting hot solution was filtered through a small plug (2.0 cm × 3.5 cm) of Celite. The bright yellow solution was concentrated to dryness and kept for further use. The Celite was allowed to dry overnight and then it was washed with two (10 mL) portions of CH2Cl2. These washings were collected in the same flask containing the residue and finally, the solution was concentrated to ca. 10 mL a rotary evaporator. Then, PPh3 (188 mg, 7.18 × 10−4 mol) was added and the mixture was stirred for 1.5 h at 298 K. After this period it was filtered and the filtrate was concentrated to dryness on a rotary evaporator giving a brownish residue which was then suspended in 30 mL of acetone and treated with LiCl (32 mg, 7.18 × 10−4 mol) and the resulting suspension was stirred at 298 K for 2.5 h. After removing the undissolved materials by filtration, the filtrate was concentrated to dryness on a rotary evaporator and dried in vacuum overnight. The yellowish-brown residue was dissolved in 10 mL of CH2Cl2 and passed through a SiO2 column (3.0 cm × 9.0 cm) using CH2Cl2 as eluant. This produced the release of two yellow bands that gave after concentration to dryness [PdCl2(PPh3)2] and compound 9 respectively. (Yield: 537 mg, 91%).). 1H–NMR data: δ = 3.90 (s, 2H, H2Fc and H5Fc), 4.02 (s, 1H, H3Fcand H5Fc), 4.03 (s, 5H, Cp), 6.02 (s, 2H, –CH2–), 6.35 (td, 1H, J = 7.5 and 1.5, H5a), 6.43 (this resonance was partially masked by the signal due to H4, 1H, H3a), 6.44 (s, 1H, H4), 6.85 (td, 1H, J = 7.5 and 1.5, H4”), 7.35–8.01 (br m, 21H, H6a, H2b–H6b and aromatic protons of the PPh3 ligand). 31P{1H} NMR data: δ 46.1. IR (solid, cm−1) 1618(w), 1588(w), 1546(w), 1479(s), 1436(s), 1419(w), 1383(w), 1185(w), 1096(s), 1035(m), 812(w), 785(s), 696(s), 532(s) and 515(m) cm−1. UV-vis. data : λ in nm (ε in M−1 cm2) =244 (49550), 268 (46000), 336 (6264). Rf(CH2Cl2) = 0.44.MS (ESI+): m/z = 785.1 {[M]–Cl–(CH2Cl2)}+. Elemental analysis calc. for C44H36N2PClFePd·CH2Cl2: C 59.63, H 4.23, N 3.09; found: C 59.5, H 4.3, N 3.0.
[Pd{1-[(η5–C5H4)Fe(η5–C5H4)–CH2]-3,5-Ph2-1-(C3HN2)}Cl(PPh3)] (10). A mixture containing compound 2 (250 mg, 5.98 × 10−4 mol), Na2[PdCl4] (176 mg, 5.98 × 10−4 mol) NaOAc·3H2O (81 mg, 5.98 × 10−4 mol) and 30 mL of methanol was stirred for 24 h at 298 K. After this period the deep-orange precipitate formed was filtered out and dried in vacuum for two days. Then it was dissolved in 20 mL of CH2Cl2, treated with PPh3 (157 mg, 5.98 × 10−4 mol) and stirred for 1.5 h at 298 K. After this period, undissolved materials were removed by filtration and discarded and the filtrate was concentrated to dryness on a rotary evaporator giving a yellowish-brown residue which was then dissolved in minimum amount of CH2Cl2 and passed through a SiO2 column (2.0 cm × 8.5 cm) using CH2Cl2 as eluant. The deep orange band was collected and concentrated to dryness on a rotary evaporator giving 10 as a deep orange microcrystalline material. This solid was collected, air-dried and then dried in vacuum for 3 days (Yield: 270 mg, 55%). 1H–NMR data: δ 3.12 (s, 1H, H2'Fc); 3.45 (s, 1H, H4'Fc), 3.60 (s, 1H, H4'Fc), 3.82 (s, 1H, H5'Fc), 3.90, 4.05 and 4.12 (s, relative intensities 2
:1:1, 4H, H2Fc–H5Fc), 4.50 (br. 1H, –CH2–), 5.20 (br. 1H, –CH2–), 6.85 (s, 1H, H4) and 7.40–8.30 (br.m. 25H, H2a–H6a, H2b–H6b and aromatic protons of the PPh3 ligand). 31P{1H} NMR data: δ 38.1. IR (solid, cm−1) 1551(w), 1479(m), 1465 (m) 1433(s) 1377 (w) 1307(m) 1181(w), 1152(w) 1095(m), 1027(m), 866(w), 820 (m), 756(s), 692(s), 534(s),517 (m) 495(m) and 489 (m) cm−1. UV-vis. data: λ in nm (ε in M−1 cm2) = 243 (49450), 255 (45986), 316 (7492), 393 (3390), and 469 (1329). Rf (CH2Cl2) = 0.35. MS (ESI+): m/z = 785.1 {[M]–Cl}+. Elemental analysis calc. for C44H36N2PClFePd:C 64.33, H, 4.39, N, 3.41 found C 64.24, H 4.61, N 3.33.
Crystallography
A prismatic crystal of compounds 2, 4, 7, 9 and 10 (sizes in Table 6), was selected and mounted on a MAR345 diffractometer with image plate detector. Unit-cell parameters were determined from 1237 (for 2), 1203 (for 4), 7287 (for 7), 217 (for 9) and 257 (for 10) reflections (in the range 3° < θ < 31°) and refined by the least-squares method. Intensities were collected with graphite monochromatized Mo-Kα radiation.| 2 | 4 | 7 | 9 | 10 | |
|---|---|---|---|---|---|
| Empirical formula | C26H22FeN2 | C28H28Cl2FeN2OPtS | C28H28Cl2FeN2OPdS | C44H36ClFeN2PPt·CH2Cl2 | C44H36ClFeN2PPd |
| Formula weight | 418.31 | 762.42 | 673.74 | 906.34 | 821.42 |
| Crystal size/mm× mm× mm | 0.2 × 0.09 × 0.09 | 0.2 × 0.1 × 0.1 | 0.2 × 0.1 × 0.1 | 0.2 × 0.1 × 0.1 | 0.1 × 0.09 × 0.09 |
| Crystal system | Triclinic | Orthorhombic | Orthorhombic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
Pbca | Pbca |
P
|
P
|
| a/Å | 10.345(7) | 15.865(4) | 15.938(5) | 9,817(6) | 10.625(6) |
| b/Å | 10.589(6) | 13.213(6) | 13.226(5) | 11.219(5) | 12.789(5) |
| c/Å | 11.221(4) | 26.348(10) | 26.424(4) | 19.794(9) | 14.469(6) |
| α (°). | 67.47(4) | 90.0 | 90.0 | 76.47(3) | 93.58(2) |
| β (°). | 67.79(4) | 90.0 | 90.0 | 82.46(3) | 105.398(2) |
| γ (°). | 82.84(5) | 90.0 | 90.0 | 69.94(3) | 107.09(2) |
| T/K | 293(2) | 173(2) | 293(2) | 293(2) | 233(2) |
| λ/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| V/Å3 | 1050.8(10) | 5523(4) | 5570(3) | 1987.9(18) | 1790.71(14) |
| Z | 2 | 8 | 8 | 2 | 2 |
| D calc./Mg × m-3 | 1.322 | 1.834 | 1.607 | 1.514 | 1.523 |
| μ/mm−1 | 0.731 | 5.879 | 1.458 | 1.094 | 1.062 |
| F(000) | 436 | 2976 | 2720 | 920 | 836 |
| Θ range for data collection/deg. | from 2.66 to 32.98 | from 2.57 to 32.53 | from 2.98 to 32.39 | from 2.59 to 32.58 | from 2.87 to 32.12 |
| N. of collected reflections | 10793 | 38751 | 40442 | 18781 | 16450 |
| N. of unique reflections {R(int)} | 5687{0.0606} | 7641{0.0833} | 7038{0.0542} | 10074{0.0481} | 8927{0.0502} |
| N. of parameters | 263 | 327 | 326 | 478 | 452 |
| Completeness to Θ = 25° | 93.0% | 96.3% | 90.2% | 92.5% | 92.4% |
| Absorption correction | Empirical | Empirical | Empirical | Empirical | Empirical |
| Max. and min transmission | 0.94 and 0.92 | 0.55 and 0.49 | 0.86 and 0.83 | 0.89 and 0.88 | 0.91 and 0.90 |
| Goodness of fit on F2 | 1.130 | 1.083 | 1.082 | 1.078 | 1.089 |
| R indices {I > 2σ(I)} | R 1 = 0.0568, | R 1 = 0.0414, | R 1 = 0.0675, | R 1 = 0.0401 | R 1 = 0.0624 |
| wR2 = 0.1842 | wR2 = 0.1174 | wR2 = 0.2041 | wR2 = 0.0995 | wR2 = 0.1884 | |
| R indices (all data) | R 1 = 0.0827, | R 1 = 0.0440, | R 1 = 0.0761, | R 1 = 0.0502 | R 1 = 0.0763 |
| wR2 = 0.1949 | wR2 = 0.1148 | wR2 = 0.2098 | wR2 = 0.1034 | wR2 = 0.1198 |
Crystal structures were solved by direct methods (for 2, 7 and 9) or by Patterson synthesis (for 4 and 10), using SHELXS computer program42 and refined by full-matrix least-squares method with the SHELX97 computer program.43 Lorentz–polarization corrections were made in all cases and absorption corrections were also carried out for 2,4 and 10.
For the five compounds, all hydrogen atoms were computed and refined using a riding model with an isotropic temperature factor equal to 1.2 times the equivalent temperature factor of the atom to which is linked. The final R(on F) and wR(on │F│2) factors were 0.057 and 0.194, respectively (for 2); 0.041 and 0.115 (for 4); 0.067 and 0.204 (for 7); 0.040 and 0.099 (for 9) and goodnesses of fit = 1.130, 1.083, 1.082, 1.078 and 1.088 (for 2, 4,7, 9 and 10, respectively) for all the observed reflections. The number of parameters and other relevant data concerning the resolution and refinement of these structures is presented in Table 6.
The crystallographic information files of the crystal structures of compounds 2, 4, 7, 9 and 10 have been deposited at the Cambridge Crystallographic Data Centre (deposition numbers: CCDC 826854–826858).
Computational details
The conformational map has been searched at the molecular mechanics level, using the augmented MM3 method39 as implemented in Cache.44 The two dihedral angles {Φ(1) and Φ(2)} have been sampled every 10 degrees, and the remaining geometric parameters have been fully optimized. DFT calculations have been performed at the B3LYP level45 using the Gaussian 03 software.38 The basis set has been chosen as follows: LANL2DZ46 for Pd, Fe, S and Cl, including polarization functions for S and Cl,47 6-31G48 for hydrogen, and 6-31G(d) including polarization functions48,49 for the remaining atoms. Solvent effects have been calculated using the C-PCM model.50Acknowledgements
This work was suported by the Ministerio de Ciencia e Innovación of Spain {grant number CTQ2009-1150, (subprogram BQU)}, FEDER Funds and by the Generalitat de Catalunya (grant number 2009-SGR-1111).References
- (a) Ferrocenes. Homogeneous Catalysis, Organic Synthesis. Materials Science, (ed) A. Togni and T. Hayashi, VCH, Weinheim (Germany), 1995 Search PubMed; (b) Ferrocene. Ligands, Materials and Biomolecules, (ed.) P. Stepnicka, Wiley, Weinheim (Germany), 2008 Search PubMed.
- (a) For recent reviews on the properties or utility of ferrocene heterocycles, see: L. V. Snegur, Y. S. Nekrasov, N. S. Sergeeva, Z. V. Zhilina, V. V. Gumenyuk, Z. A. Starikova, A. A. Simenel, N. B. Morozova, I. K. Sviridova and V. N. Babin, Appl. Organomet. Chem., 2008, 22, 139–147 CrossRef CAS; (b) G. Fu, Asymm. Synth., 2007, 186–90 CAS; (c) J. Yoon, S. K. Kim, N. J. Singh and K. S. Kim, Chem. Soc. Rev., 2006, 35, 355–360 RSC; (d) R. Gómez Arrayas, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674–7715 CrossRef; (e) D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5985 CrossRef CAS.
- (a) For recent advances on this field, see: M. E. Weiss, D. F. Fischer, Z. Xin, S. Jautze, W. B. Schweizer and R. Peters, Angew. Chem., Int. Ed., 2006, 45, 5694–5698 CrossRef CAS; (b) C. E. Anderson, Y. Donde, C. J. Douglas and L. E. Overman, J. Org. Chem., 2005, 70, 648–657 CrossRef CAS; (c) D. Pou, A. E. Platero-Prats, S. Pérez, C. López, X. Solans, M. Font-Bardía, P. W. N. M. van Leeuwen, G. P. F. van Strijdonck and Z. Freixa, J. Organomet. Chem., 2007, 692, 5017–5025 CrossRef CAS; (d) S. Pérez, C. López, A. Caubet, X. Solans, M. Font-Bardía, A. Roig and E. Molins, Organometallics, 2006, 25, 596–601 CrossRef; (e) S. Jautze, S. Diethelm, W. Frey and R. Peters, Organometallics, 2009, 28, 2001–2004 CrossRef CAS; (f) T. Mochida, H. Shimizu, K. Ozakawa, F. Sato and D. Kuwahara, J. Organomet. Chem., 2007, 692, 1834–1844 CrossRef CAS; (g) R. Peters, D. F. Fischer, S. Jautze, F. M. Koch, T. Kull, P. S. Tiseni, Z. Xin and M. Zajac, Chimia, 2008, 62, 497–505 CrossRef CAS.
- (a) A. Moyano, M. Rosol, R. M. Moreno, C. López and M. A. Maestro, Angew. Chem., Int. Ed., 2005, 44, 1865–1869 CrossRef CAS; (b) A. González, C. López, X. Solans, M. Font-Bardía and E. Molins, J. Organomet. Chem., 2008, 693, 2119–2131 CrossRef; (c) P. K. Basu, A. González, C. López, M. Font-Bardía and T. Calvet, J. Organomet. Chem., 2009, 694, 3633–3642 CrossRef CAS.
- (a) J. J. Li and G. W. Gribble, Palladium in Heterocyclic Chemistry, Pergamon, 2000, New York (USA) Search PubMed.
- (a) V. Montoya, J. Pons, X. Solans, M. Font-Bardía and J. Ros, Inorg. Chim. Acta, 2005, 358, 2312–2318 CrossRef CAS; (b) V. Montoya, J. Pons, X. Solans, M. Font-Bardía and J. Ros, Inorg. Chim. Acta, 2005, 358, 2763–2769 CrossRef CAS; (c) J. Chakraborty, M. K. Saha and P. Benerjee, Inorg. Chem. Commun., 2007, 10, 671–676 CrossRef CAS.
- (a) K. Li, M. S. Mohlala, T. V. Segapelo, P. M. Shumbula, I. A. Guzei and J. Darkwa, Polyhedron, 2008, 27, 1017–1023 CrossRef CAS; (b) R. Y. Mawo, D. M. Johnson, J. L. Wood and I. P. Smoliakova, J. Organomet. Chem., 2008, 693, 33–45 CrossRef CAS; (c) I. Ara, L. R. Falvello, J. Fornies, R. Lasheras, A. Martin, O. Oliva and V. Sicilia, Inorg. Chim. Acta, 2006, 359, 4574–4584 CrossRef CAS; (d) I. Ara, J. Forniés, R. Lasheras, A. Martin and V. Sicilia, Eur. J. Inorg. Chem., 2006, 948–957 CrossRef CAS; (e) Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 6581–6585 CrossRef CAS; (f) F. Churruca, R. SanMartin, I. Tellitu and E. Domínguez, Synlett., 2005, 3116–3120 CAS; (g) F. López-Linares, O. Colmenares, E. Catari and A. Karam, React. Kinet. Catal. Lett., 2005, 85, 139–144 CrossRef; (h) A. Satake and T. Nakata, J. Am. Chem. Soc., 1998, 120, 10391–10396 CrossRef CAS; (i) E. Budzisz, U. Krajewska, M. Rozalski, A. Szulawska, M. Czyz and B. Nawrot, Eur. J. Pharmacol., 2004, 502, 59–65 CrossRef CAS; (j) M. C. Torralba, M. Cano, J. A. Campo, J. V. Heras, E. Pinilla and M. R. Torres, Inorg. Chem. Commun., 2002, 5, 887–890 CrossRef CAS.
- (a) S. Y. Chang, J-L. Chen, Y. Chi, Y-M. Cheng, G-H. Lee, C-M. Jiang and P. T. Chou, Inorg. Chem., 2007, 46, 11202–11212 CrossRef CAS; (b) A. Eisenwiener, M. Neuburger and T. A. Kaden, Dalton Trans., 2007, 36, 218–233 RSC; (c) A. V. Khripun, M. Haukka and V. Y. Kukushkin, Russ. Chem. Bull., 2006, 55, 247–255 CrossRef CAS; (d) A. V. Khripun, S. I. Selivanov, V. Y. Kukushkin and M. Haukka, Inorg. Chim. Acta, 2006, 359, 320–6 CrossRef CAS; (e) E. Ciesielska, A. Szulawska, K. Studzian, J. Ochocki, K. Malinowska, K. Kik and L. Szmigiero, J. Inorg. Biochem., 2006, 100, 1579–1585 CrossRef CAS.
- (a) For a general overview on cyclopallada- and cycloplatinated compounds their applications, see: J. Dupont and M. Pfeffer, Palladacycles. Synthesis, Characterization and Applications, Wiley-VCH, Weinheim (Germany), 2008 Search PubMed; (b) M. Ghedini, I. Aiello, A. Crispini, A. Golemme, M. La Deda and D. Pucci, Coord. Chem. Rev., 2006, 250, 1373–1390 CrossRef CAS; (c) D. A. Alonso and C. Najera, Chem. Soc. Rev., 2010, 39, 2891–2902 RSC; (d) I. Omae, J. Organomet. Chem., 2007, 692, 2608–32 CrossRef CAS; (e) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS; (f) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS.
- (a) For recent advances in pallada- and platinacycles, see: J. Albert, R. Bosque, M. Crespo, J. Granell, J. Rodriguez and J. Zafrilla, Organometallics, 2010, 29, 4619–4627 CrossRef CAS; (b) M. Crespo, T. Calvet and M. Font-Bardia, Dalton Trans., 2010, 39, 6936–6938 RSC; (c) M-Y. Yuen, S. C. F. Kui, K-H. Low, C.-C. Kwok, S. S.-Y. Chui, C.-W. Ma, N. Zhu and C.-M. Che, Chem.–Eur. J., 2010, 16, 14131–14141 CrossRef CAS; (d) C. López, S. Pérez, X. Solans, M. Font-Bardía, A. Roig, E. Molins, P. W. N. M. van Leeuwen and G. P. F. van Strijdonck, Organometallics, 2007, 26, 571–576 CrossRef; (e) T-K. Zhang, K. Yuan and X-L. Hou, J. Organomet. Chem., 2007, 692, 1912–19 CrossRef CAS; (f) P. G. Evans, N. A. Brown, G. J. Clarkson, C. P. Newman and J. P. Rourke, J. Organomet. Chem., 2006, 691, 1251–1256 CrossRef CAS.
- (a) For applications of palladacycles in homogeneous catalysis, see: A. S. Gruber, D. Zim, G. Ebeling, A. L. Monteiro and J. Dupont, Org. Lett., 2000, 2, 1287–1290 CrossRef CAS; (b) D. Zim, A. S. Gruber, G. Ebeling, J. Dupont and A. L. Monteiro, Org. Lett., 2000, 2, 2881–2884 CrossRef CAS; (c) J. Dupont, M. Pfeffer and J. Spencer, Eur. J. Inorg. Chem., 2001, 1917–1927 CrossRef CAS; (d) I. Moreno, R. San Martin, M. T. Herrero and E. Dominguez, Curr.Topics Catal., 2009, 8, 91–102 CAS and references cited in these articles..
- (a) For examples of the antitumoral activity of this sort of compounds, see: G. Xu, Z. Yan, N. Wang and Z. Liu, Eur. J. Med. Chem., 2011, 46, 356–363 CrossRef CAS; (b) L. Ronconi and P. J. Sadler, Dalton Trans., 2011, 40, 262–268 RSC; (c) J. Aleman, V. del Solar, L. Cubo, A. G. Quiroga and C. Navarro-Ranninger, Dalton Trans., 2010, 39, 10601–10607 RSC; (d) R. W-Y. Sun, D. L. Ma, E. L-M. Wong and C-M. Che, Dalton Trans., 2007, 35, 4884–4892 Search PubMed; (e) A. P. Neves, G. B. da Silva, M. D. Vargas, C. B. Pinheiro, L. C. Visentin, J. D. B. M. Filho, A. J. Araujo, L. V. Costa-Lotufo, C. Pessoa and M. O. de Moraes, Dalton Trans., 2010, 39, 10203–10216 RSC; (f) G. Wagner, A. Marchant and J. Sayer, Dalton Trans., 2010, 39, 7747–7759 RSC; (g) N. Margiotta, N. Denora, R. Ostuni, V. Laquintana, A. Anderson, S. W. Johnson, G. Trapani and G. Natile, J. Med. Chem., 2010, 53, 5144–5154 CrossRef CAS; (h) J. S. Saad, M. Benedetti, G. Natile and L. G. Marzilli, Inorg. Chem., 2010, 49, 5573–5583 CrossRef CAS; (i) K. L. Ciesienski, L. M. Hyman, D. T. Yang, K. L. Haas, M. G. Dickens, R. J. Holbrook and K. J. Franz, Eur. J. Inorg. Chem., 2010, 2224–2228 CrossRef CAS; (j) Y. Y. Scaffidi-Domianello, K. Meelich, M. A. Jakupec, V. B. Arion, V. Y. Kukushkin, M. Galanski and B. K. Keppler, Inorg. Chem., 2010, 49, 5669–5678 CrossRef CAS; (k) F. J. Ramos-Lima, V. Moneo, A. G. Quiroga, A. Carnero and C. Navarro-Ranninger, Eur. J. Med. Chem., 2010, 45, 134–141 CrossRef CAS.
- (a) For macromolecules containing cyclopalladated or cycloplatinated units: M. López-Torres, A. Fernández, J. J. Fernández, A. Suárez, S. Castro-Juiz, J. M. Vila and M. T. Pereira, Organometallics, 2001, 20, 1350–1353 CrossRef; (b) C. López, A. Caubet, S. Pérez, X. Solans and M. Font-Bardía, J. Organomet. Chem., 2003, 681, 82–90 CrossRef; (c) A. Fernández, E. Pereira, J. J. Fernández, M. López-Torres, A. Suarez, R. Mosteiro, M. T. Pereira and J. M. Vila, New J. Chem., 2002, 26, 895–901 RSC; (d) I. Aiello, U. Caruso, M. Ghedini, B. Panunzi, A. Quatela, A. Roviello and F. Sarcinelli, Polymer, 2003, 44, 7635–7643 CrossRef CAS.
- (a) For examples of their utility in organic and organometallic synthesis, see: X-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047–9050 CrossRef CAS; (b) A. Dumrath, X-F. Wu, H. Neumann, A. Spannenberg, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8988–8992 CrossRef CAS; (c) A. Sivaramakrishna, H. S. Clayton, M. M. Mogorosi and J. R. Moss, Coord. Chem. Rev., 2010, 254, 2904–2932 CrossRef CAS; (d) E. Negishi, G. Wang, H. Rao and Z. Xu, J. Org. Chem., 2010, 75, 3151–3182 CrossRef CAS; (e) M-J. Jin and D-H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119–1122 CrossRef CAS.
- Y. Donde and L. E. Overman, J. Am. Chem. Soc., 1999, 121, 2933–2934 CrossRef CAS.
- (a) M. D. Joksovic, V. Markovic, Z. D. Juranic, T. Stanojkovic, L. S. Jovanovic, I. S. Damljanovic, K. M. Szecsenyi, N. Todorovic, S. Trifunovic and R. D. Vukicevic, J. Organomet. Chem., 2009, 694, 3935–3942 CrossRef CAS; (b) I. Damiljanovic, M. Vukicevic, N. Radulovic, R. Palic, E. Ellmerer, Z. Ratkovic, M. D. Joksovic and R. D. Vukicevic, Bioorg. Med. Chem. Lett., 2009, 19, 1093–1096 CrossRef; (c) M. Zora and M. Goermen, J. Organomet. Chem., 2007, 692, 5026–5032 CrossRef CAS; (d) M. Joksovic, Z. Ratkovic, M. Vukicevic and R. D. Vukicevic, SynLett., 2006, 2581–2584 CAS; (e) J. L. Delgado, M. E. El-Khouly, Y. Araki, M. J. Gomez-Escalonilla, P. De la Cruz, F. Oswald, O. Ito and F. Langa, Phys. Chem. Chem. Phys., 2006, 8, 4104–4111 RSC; (f) W-Q. Shen, Y-J. Wang, K-L. Chen, G-H. Lee and C. K. Lai, Tetrahedron, 2006, 62, 8035–8044 CrossRef CAS; (g) A. H. Ilkhechi, M. Bolte, H-W. Lerner and M. Wagner, J. Organomet. Chem., 2005, 690, 1971–1977 CrossRef CAS; (h) U. Burckhardt, D. Drommi and A. Togni, Inorg. Chim. Acta, 1999, 296, 183–194 CrossRef CAS.
- (a) Y-N. Liu, G. Orlowski, G. Schatte and H-B. Kraatz, Inorg. Chim. Acta, 2005, 358, 1151–61 CrossRef CAS; (b) G. L. G. Effendy, M. Pellei, C. Pedinari, C. Santini, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2001, 315, 153–62 CrossRef CAS.
- (a) H. Qian, X. Cui, M. Tang, C. Liu, C. Liu and Y. Wu, New J. Chem., 2009, 33, 668–674 RSC; (b) L. L. Troitskaya, Z. A. Starikova, T. V. Desmeshchik, S. T. Ovseenko, E. V. Vorontsov and V. I. Sokolov, J. Organomet. Chem., 2005, 690, 3976–3982 CrossRef CAS.
- (a) A. Zucca, G. L. Petretto, S. Stoccoro, M. A. Cinellu, G. Minghetti, M. Manassero, C. Manassero, L. Male and A. Albinati, Organometallics, 2006, 25, 2253–2265 CrossRef CAS; (b) I. Moreno, R. SanMartin, B. Ines, M. T. Herrero and E. Dominguez, Curr. Org. Chem., 2009, 13, 878–895 CrossRef CAS.
- (a) V. I. Boev, L. V. Snegur, V. N. Babin and Y. S. Nekrasov, Russ. Chem. Rev., 1997, 66, 613–636 CrossRef; (b) K. Pagel, A. Werner and W. Friedrichsen, J. Organomet. Chem., 1994, 481, 109–123 CrossRef CAS.
- A. González, J. Marquet and M. Moreno-Mañas, Tetrahedron, 1986, 42, 4253–4257 CrossRef.
- M. Rosemblum and W. G. Howels, J. Am. Chem. Soc., 1962, 84, 1167–1172 CrossRef.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, B58, 380–388 CrossRef CAS.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, Inorg. Chem., 1972, 11, 1280–4 CrossRef CAS.
- (a) S. Pérez, C. López, A. Caubet, X. Solans and M. Font-Bardía, Eur. J. Inorg. Chem., 2008, 1599–1612 CrossRef; (b) C. López, A. Caubet, S. Pérez, X. Solans and M. Font-Bardía, Chem. Commun., 2004, 540–541 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th ed., Wiley, New York, USA, 1997 Search PubMed.
- (a) E. Szłyk, I. Lakomska, A. Surdykowski, T. Glowiak, L. Pazderski, J. Sitkowski and L. Kokerski, Inorg. Chim. Acta, 2002, 333, 93–99 CrossRef; (b) N. Nédélec and F. D. Rochon, Inorg. Chem., 2001, 40, 5236–5244 CrossRef; (c) N. Nédélec and F. D. Rochon, Inorg. Chim. Acta, 2001, 319, 95–98 CrossRef.
- (a) B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- (a) For compounds with N-donor heterocyclic ligands see for instance: A. Cornia, A. C. Fabretti, M. Bonivento and L. Cattalini, Inorg. Chim. Acta, 1997, 255, 405–409 CrossRef CAS; (b) R. Melanson and F. D. Rochon, Inorg. Chem., 1978, 17, 679–681 CrossRef CAS.
- (a) X. Riera, C. López, A. Caubet, V. Moreno, X. Solans and M. Font-Bardía, Eur. J. Inorg. Chem., 2001, 2135–2141 CrossRef CAS; (b) R. Martin, M. Crespo, M. Font-Bardia and T. Calvet, Organometallics, 2009, 28, 587–597 CrossRef CAS; (c) J. Rodríguez, J. Zafrilla, J. Albert, M. Crespo, J. Granell, T. Calvet and M. Font-Bardia, J. Organomet. Chem., 2009, 694, 2467–2475 CrossRef.
- J. Vicente, J. A. Abad, A. D. Frankland and M. C. Ramírez de Arellano, Chem.–Eur. J., 1999, 5, 3066–3075 CrossRef CAS.
- I. Lakomska, M. Barwiolek, A. Wojtczak and E. Szlyk, Polyhedron, 2007, 26, 5349–5354 CrossRef CAS.
- (a) C. López, J. Sales, X. Solans and R. Zquiak, J. Chem. Soc., Dalton Trans., 1992, 2321–2328 RSC; (b) R. Bosque, C. López, J. Sales, X. Solans and M. Font-Bardía, J. Chem. Soc., Dalton Trans., 1994, 735–745 RSC.
- Y. Wu, S. Huo, J. Gong, X. Cui, L. Ding, K. Ding, C. Du, Y. Liu and M. Song, J. Organomet. Chem., 2001, 637-639, 27–46 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) A. D. Ryabov, S. Otto, P. V. Samuleev, V. A. Polyakov, L. Alexandrova, G. M. Kazankov, S. Shova, M. Revenko, J. Lipkowski and M. H. Johansson, Inorg. Chem., 2002, 41, 4286–4294 CrossRef CAS; (b) S. Otto, A. Chanda, P. V. Samuleev and A. D. Ryabov, Eur. J. Inorg. Chem., 2006, 2561–2566 CrossRef CAS.
- R. Bosque and F. Maseras, Eur. J. Inorg. Chem., 2005, 4040–4047 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R .L. Martin, D.J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian 03 (Revision C.02). Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- N. L. Allinger, Y. H. Yuh and J. H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- Z. Szafran, R. M. Pike and M. M. Singh, Microscale Inorganic Chemistry. A Comprehensive Laboratory ExperienceJohn Wiley & Sons, New York (USA), 1991, p. 218 Search PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 4th Ed. Butterworth–Heinemann, 1996, Oxford (UK) Search PubMed.
- G. M. Sheldrick, SHELXS A program for automatic solution of crystal structure refinement, 1997, Univ. Goettingen, (Germany) Search PubMed.
- G. M. Sheldrick, SHELX97A program for crystal structure refinement, 1997, Univ. Goettingen, (Germany) Search PubMed.
- CacheWork System Pro Version 7.5.085 UserGuide, Fujitsu, Beaverton, Oregon, USA, 2007.
- (a) A. D.Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- (a) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- A. Höllwarth, M. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comp. Chem., 2003, 24, 669–681 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Conformational map showing the variation of the total energy of the ligand as a function of the torsion angles Φ(1) and Φ(2) (Fig. S1) and tables containing final atomic coordinates of the optimized geometries of the three types of complexes (A–C, shown in the lower part of Table S5) with L = PH3, dmso or PPh3 used as models in the DFT study together with their calculated total energy in vacuum, in toluene and in methanol (Tables S1-S9). CCDC reference numbers 826854–826858. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra01080h |
| ‡ The separation between the centroid of the ring defined by the set of atoms [C(6)–C(10] of a molecule at (x, y, z) and the H(18) atom of a close one located at (−x, −y, 1 − z) is 2.997 Å and the distance between the center of the ring [C(6)–C(10)] of the unit at (x, y, z) and the H(24) of another one at (2 − x, 1 − y, −z) is 3.036 Å. The separation between the H(16) atom and the phenyl ring [C(21)–C(26)] of another unit is 3.963 Å. |
| § For illustrative purposes, the yields of 5 for different reaction periods (t) are given: 67.8, 69.1 and 70.2% for t = 3.5, 12 and 24 h, respectively. |
| ¶ In 2, the main plane of the heterocycle forms angles of 80.9° and 80.3° with the two pentagonal rings of the Fc unit. |
| || The success of all preparations described in this section is strongly dependent on the quality of the methanol used. The presence of small amounts of water reduces significantly the yield of the process. Thus the use of high quality (HPLC-grade) methanol is strongly recommended. In all cases the reaction flask was protected from the light with aluminium foil. |
| This journal is © The Royal Society of Chemistry 2012 |